Department of Mechanics, Shanghai University, Shanghai, 200444, China.
Shanghai Institute of Applied Mathematics and Mechanics, Shanghai University, Shanghai, 200444, China.
Sci Rep. 2017 Jul 12;7(1):5167. doi: 10.1038/s41598-017-05366-1.
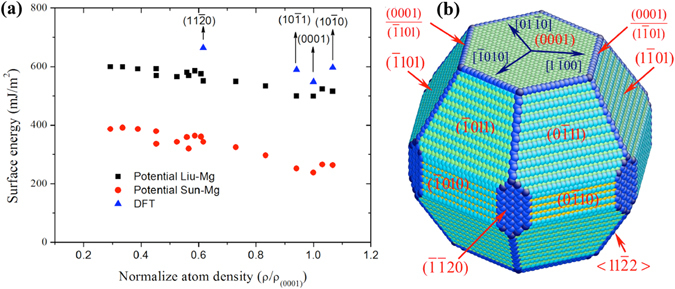
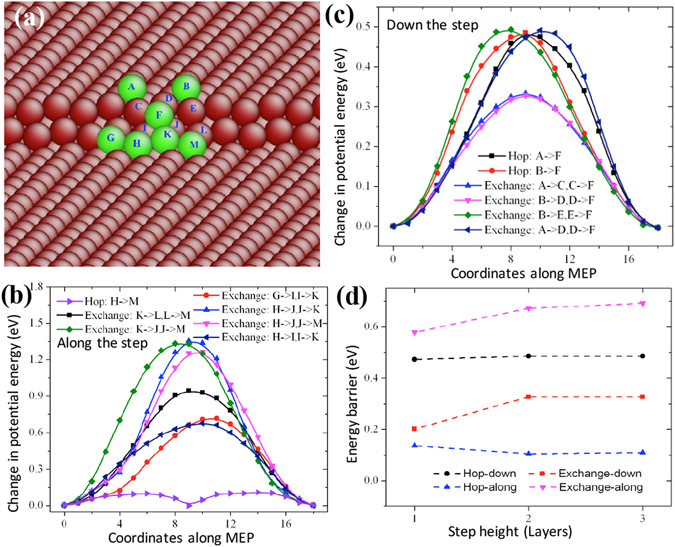
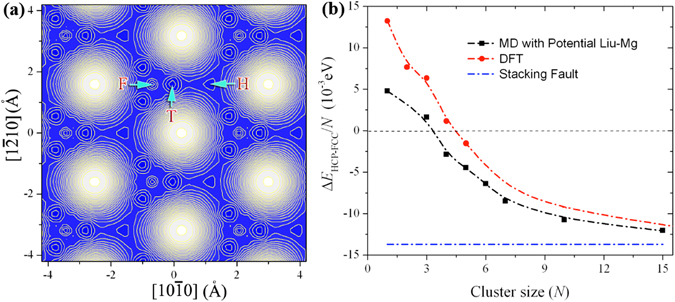
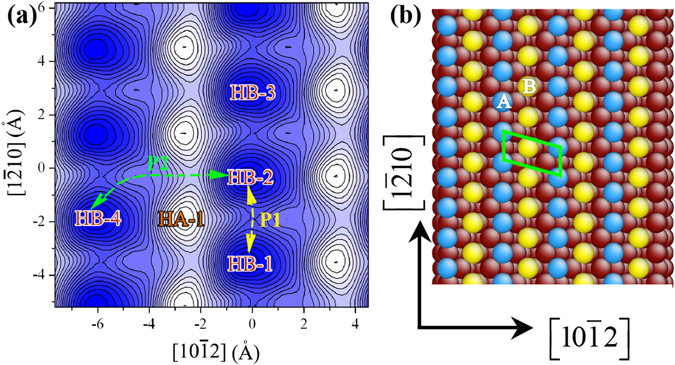
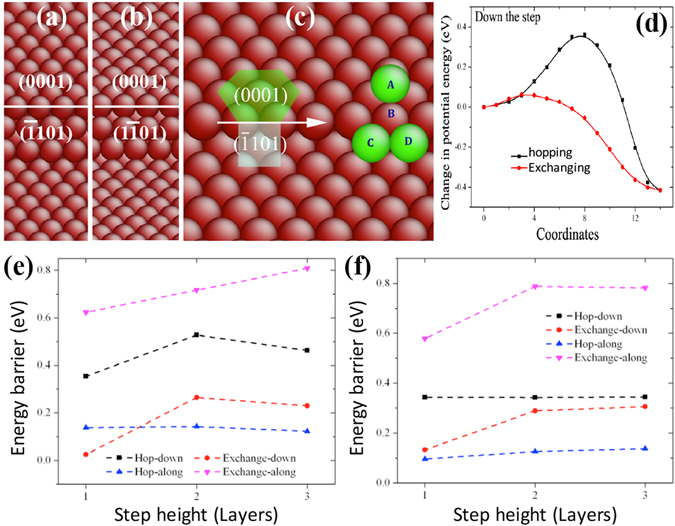
The formation and diffusion energies of atomic clusters on Mg surfaces determine the surface roughness and formation of faulted structure, which in turn affect the mechanical deformation of Mg. This paper reports first principles density function theory (DFT) based quantum mechanics calculation results of atomic clustering on the low energy surfaces {0001} and [Formula: see text]. In parallel, molecular statics calculations serve to test the validity of two interatomic potentials and to extend the scope of the DFT studies. On a {0001} surface, a compact cluster consisting of few than three atoms energetically prefers a face-centered-cubic stacking, to serve as a nucleus of stacking fault. On a [Formula: see text], clusters of any size always prefer hexagonal-close-packed stacking. Adatom diffusion on surface [Formula: see text] is high anisotropic while isotropic on surface (0001). Three-dimensional Ehrlich-Schwoebel barriers converge as the step height is three atomic layers or thicker. Adatom diffusion along steps is via hopping mechanism, and that down steps is via exchange mechanism.
镁表面原子团簇的形成和扩散能决定了表面粗糙度和位错结构的形成,从而影响镁的机械变形。本文报道了基于第一性原理密度泛函理论(DFT)的镁低能表面{0001}和[Formula: see text]上原子团簇的量子力学计算结果。同时,分子静力学计算用于检验两种原子间势的有效性,并扩展 DFT 研究的范围。在{0001}表面上,由少于三个原子组成的紧密团簇在能量上优先采用面心立方堆积,作为堆垛层错的核心。在[Formula: see text]表面上,任何大小的团簇总是优先采用六方密堆积堆积。表面[Formula: see text]上的原子扩散具有各向异性,而表面(0001)上的原子扩散具有各向同性。当台阶高度为三个原子层或更厚时,三维 Ehrlich-Schwoebel 势垒收敛。原子沿着台阶的扩散是通过跃迁机制,而沿着台阶的扩散是通过交换机制。