John Innes Centre, Norwich, UK.
BMC Plant Biol. 2018 Jan 25;18(1):22. doi: 10.1186/s12870-018-1241-5.
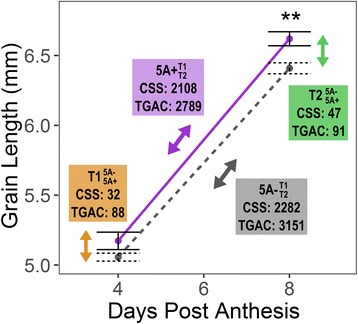
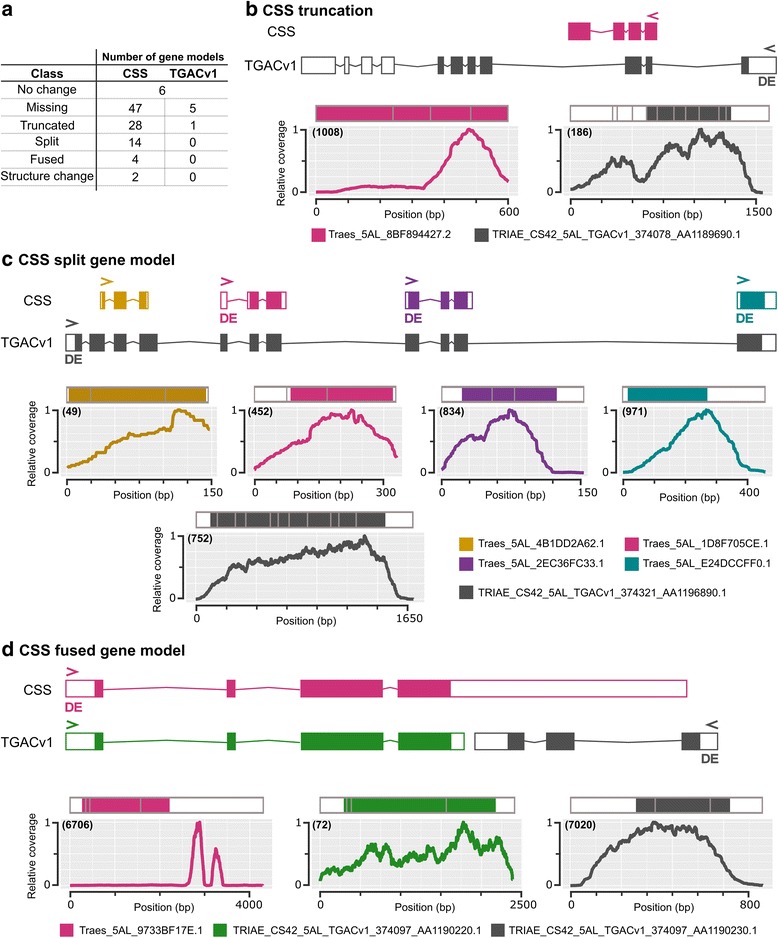
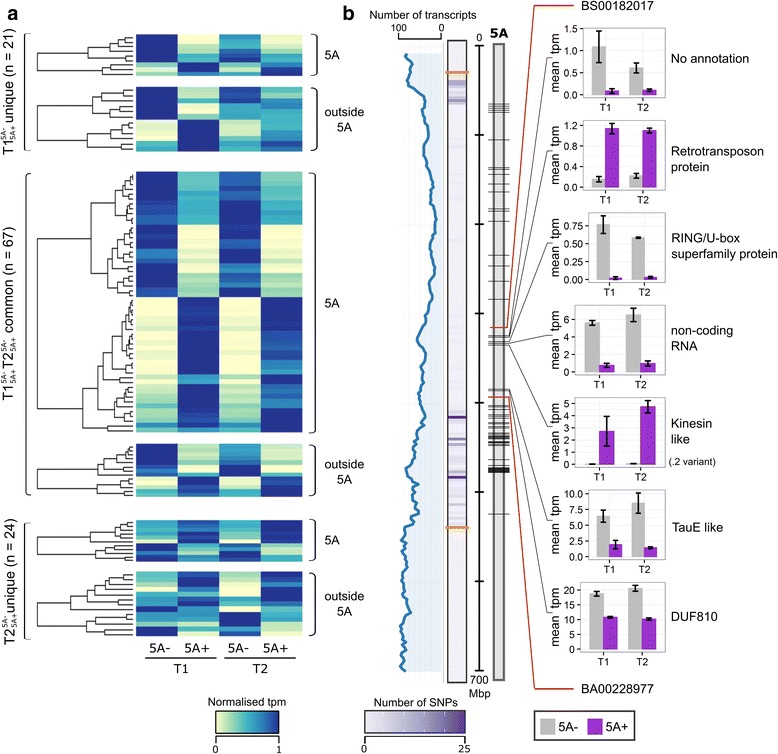
There is an urgent need to increase global crop production. Identifying and combining specific genes controlling distinct biological processes holds the potential to enhance crop yields. Transcriptomics is a powerful tool to gain insights into the complex gene regulatory networks that underlie such traits, but relies on the availability of a high-quality reference sequence and accurate gene models. Previously, we identified a grain weight QTL on wheat chromosome 5A (5A QTL) which acts during early grain development to increase grain length through cell expansion in the pericarp. In this study, we performed RNA-sequencing on near isogenic lines (NILs) segregating for the 5A QTL and used the latest gene models to identify differentially regulated genes and pathways that potentially influence pericarp cell size and grain weight in wheat.
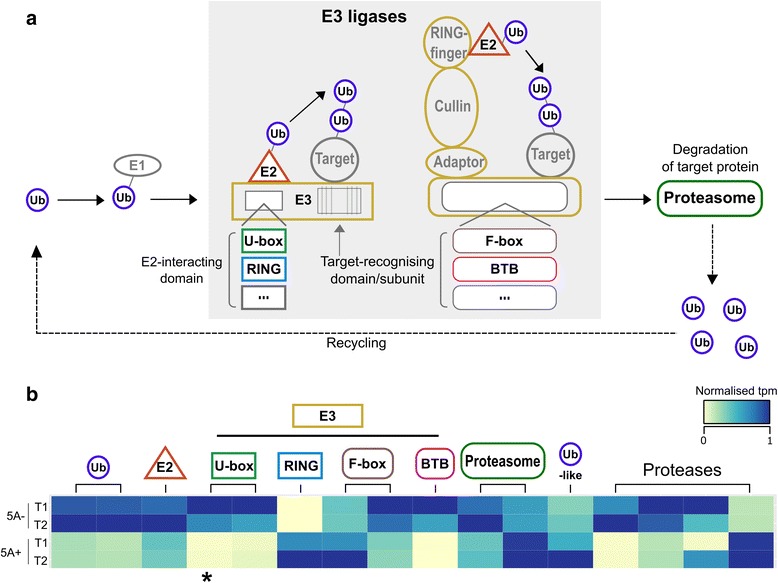
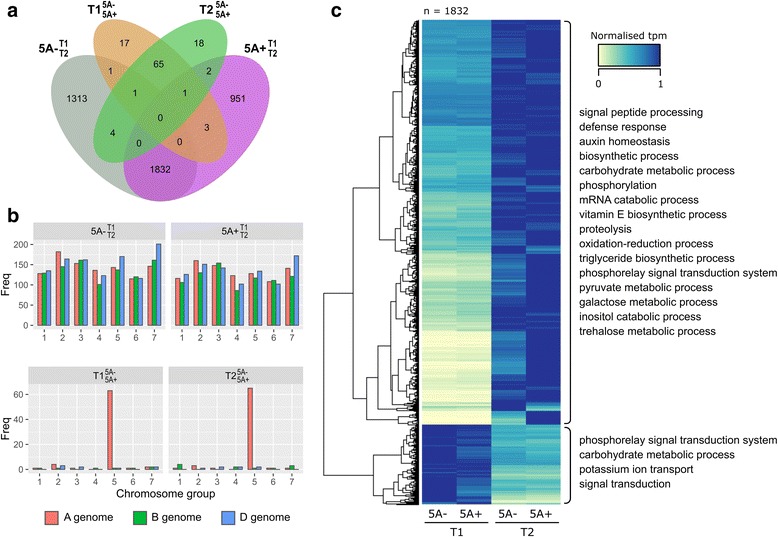
We sampled grains at 4 and 8 days post anthesis and found that genes associated with metabolism, biosynthesis, proteolysis and the defence response are upregulated during this stage of grain development in both NILs. We identified a specific set of 112 transcripts differentially expressed (DE) between 5A NILs at either time point, including eight potential candidates for the causal 5A gene and its downstream targets. The 112 DE transcripts had functional annotations including non-coding RNA, transposon-associated, cell-cycle control, ubiquitin-related, heat-shock, transcription and histone-related. Many of the genes identified belong to families that have been previously associated with seed/grain development in other species. Notably, we identified DE transcripts at almost all steps of the pathway associated with ubiquitin-mediated protein degradation. In the promoters of a subset of DE transcripts we identified enrichment of binding sites associated with C2H2, MYB/SANT, YABBY, AT-HOOK and Trihelix transcription factor families.
In this study, we identified DE transcripts with a diverse range of predicted biological functions, reflecting the complex nature of the pathways that control early grain development. Few of these are the direct orthologues of grain size genes in other species and none have been previously characterised in wheat. Further functional characterisation of these candidates and how they interact will provide novel insights into the control of grain size in cereals.
全球作物生产急需提高。鉴定和组合控制不同生物过程的特定基因有可能提高作物产量。转录组学是深入了解控制这些性状的复杂基因调控网络的有力工具,但依赖于高质量参考序列和准确基因模型的可用性。此前,我们在小麦 5A 染色体上鉴定到一个粒重 QTL(5A QTL),该 QTL 在籽粒发育早期通过果皮细胞扩展来增加籽粒长度。在这项研究中,我们对分离 5A QTL 的近等基因系(NILs)进行了 RNA-seq 测序,并使用最新的基因模型来鉴定潜在影响小麦果皮细胞大小和粒重的差异调节基因和途径。
我们在授粉后 4 天和 8 天取样籽粒,发现两个 NILs 中与代谢、生物合成、蛋白水解和防御反应相关的基因在籽粒发育的这个阶段上调。我们鉴定了一组 112 个在两个时间点的 5A NILs 之间差异表达(DE)的转录本,包括 5A 基因及其下游靶基因的 8 个潜在候选基因。112 个 DE 转录本具有非编码 RNA、转座子相关、细胞周期控制、泛素相关、热休克、转录和组蛋白相关等功能注释。鉴定到的许多基因属于已在其他物种中与种子/籽粒发育相关的家族。值得注意的是,我们在与泛素介导的蛋白降解相关的途径的几乎所有步骤中都鉴定到了 DE 转录本。在一组 DE 转录本的启动子中,我们鉴定到与 C2H2、MYB/SANT、YABBY、AT-HOOK 和三螺旋转录因子家族相关的结合位点富集。
在这项研究中,我们鉴定到具有广泛预测生物学功能的 DE 转录本,反映了控制早期籽粒发育的途径的复杂性。其中很少是其他物种中粒大小基因的直接同源物,也没有在小麦中进行过特征描述。这些候选基因的进一步功能特征及其相互作用将为谷物粒大小的控制提供新的见解。