Scala Rosa, Maqoud Fatima, Zizzo Nicola, Mele Antonietta, Camerino Giulia Maria, Zito Francesco Alfredo, Ranieri Girolamo, McClenaghan Conor, Harter Theresa M, Nichols Colin G, Tricarico Domenico
Section of Pharmacology, Department of Pharmacy-Pharmaceutical Sciences, University of Bari "Aldo Moro", Bari, Italy.
Section of Veterinary Pathology and Comparative Oncology, Department of Veterinary Medicine, University of Bari "Aldo Moro", Valenzano, Italy.
Front Pharmacol. 2020 Nov 30;11:604885. doi: 10.3389/fphar.2020.604885. eCollection 2020.
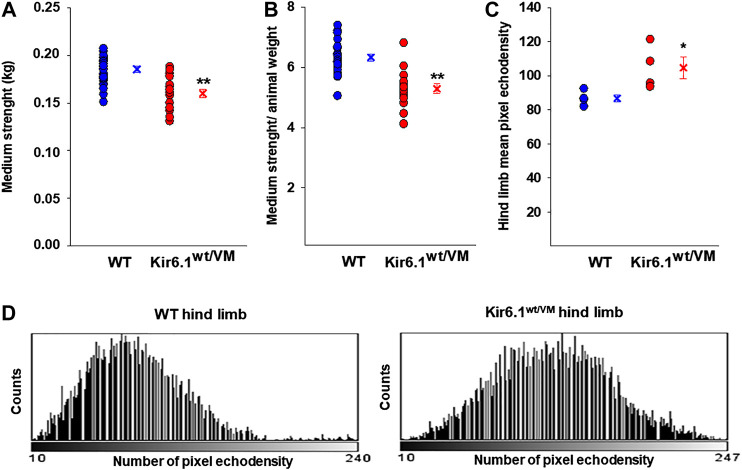
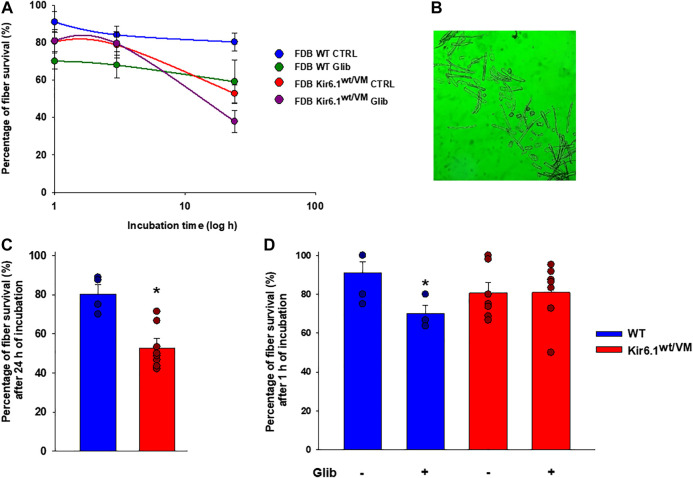
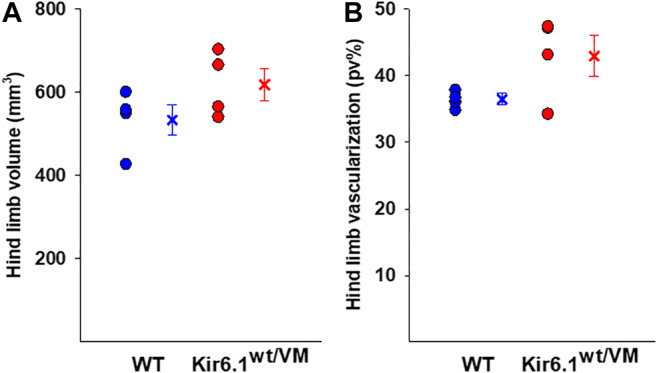
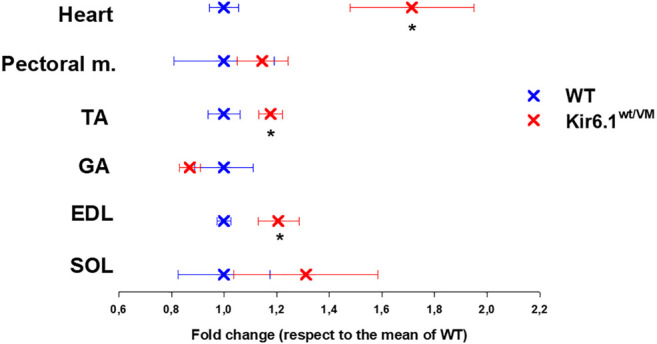
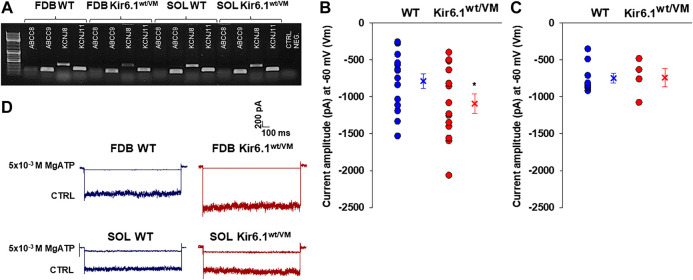
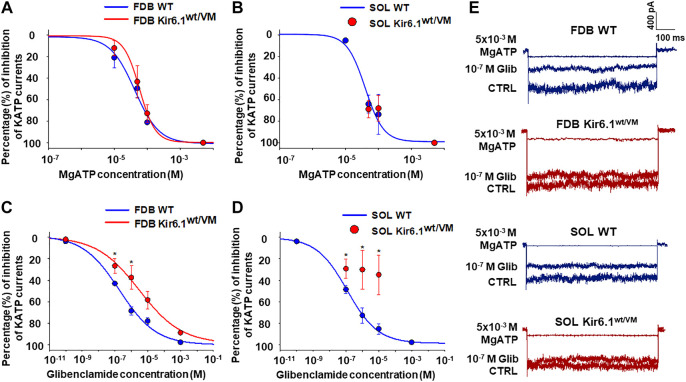
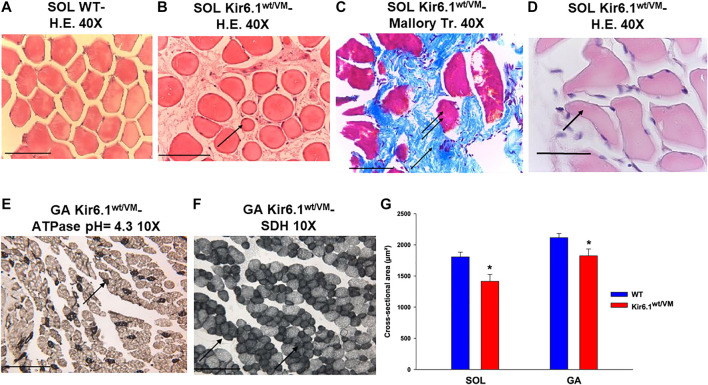
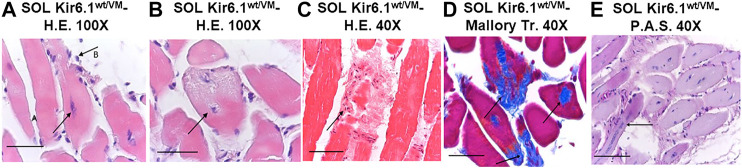
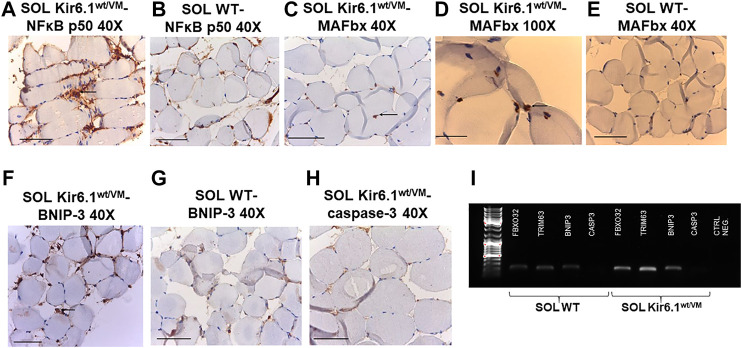
Cantù syndrome (CS) arises from mutations in and genes that lead to gain of function (GOF) of ATP-sensitive potassium (KATP) channels containing SUR2A and Kir6.1 subunits, respectively, of KATP channels. Pathological consequences of CS have been reported for cardiac and smooth muscle cells but consequences in skeletal muscle are unknown. Children with CS show muscle hypotonia and adult manifest fatigability. We analyzed muscle properties of Kir6.1[V65M] CS mice, by measurements of forelimb strength and ultrasonography of hind-limb muscles, as well as assessing KATP channel properties in native (FDB) and (SOL) fibers by the patch-clamp technique in parallel with histopathological, immunohistochemical and Polymerase Chain Reaction (PCR) analysis. Forelimb strength was lower in Kir6.1 mice than in WT mice. Also, a significant enhancement of echodensity was observed in hind-limb muscles of Kir6.1 mice relative to WT, suggesting the presence of fibrous tissue. There was a higher KATP channel current amplitude in Kir6.1 FDB fibers relative to WT and a reduced response to glibenclamide. The IC of glibenclamide to block KATP channels in FDB fibers was 1.3 ± 0.2 × 10 M in WT and 1.2 ± 0.1 × 10 M in Kir6.1 mice, respectively; and it was 1.2 ± 0.4 × 10 M in SOL WT fibers but not measurable in Kir6.1 fibers. The sensitivity of the KATP channel to MgATP was not modified in Kir6.1 fibers. Histopathological/immunohistochemical analysis of SOL revealed degeneration plus regressive-necrotic lesions with regeneration, and up-regulation of Atrogin-1, MuRF1, and BNIP3 mRNA/proteins in Kir6.1 mice. Kir6.1 mutation in skeletal muscle leads to changes of the KATP channel response to glibenclamide in FDB and SOL fibers, and it is associated with histopathological and gene expression changes in slow-twitch muscle, suggesting marked atrophy and autophagy.
坎图综合征(CS)源于 和 基因的突变,这些突变分别导致含SUR2A和Kir6.1亚基的ATP敏感性钾(KATP)通道功能获得(GOF)。CS对心脏和平滑肌细胞的病理后果已有报道,但对骨骼肌的影响尚不清楚。患有CS的儿童表现出肌张力减退,成年人则表现出易疲劳。我们通过测量前肢力量和对后肢肌肉进行超声检查,分析了Kir6.1[V65M] CS小鼠的肌肉特性,并通过膜片钳技术评估天然趾长伸肌(FDB)和比目鱼肌(SOL)纤维中的KATP通道特性,同时进行组织病理学、免疫组织化学和聚合酶链反应(PCR)分析。Kir6.1小鼠的前肢力量低于野生型小鼠。此外,相对于野生型小鼠,在Kir6.1小鼠的后肢肌肉中观察到回声密度显著增强,提示存在纤维组织。与野生型相比,Kir6.1 FDB纤维中的KATP通道电流幅度更高,对格列本脲的反应降低。格列本脲阻断FDB纤维中KATP通道的IC50在野生型小鼠中为1.3±0.2×10⁻⁶ M,在Kir6.1小鼠中为1.2±0.1×10⁻⁶ M;在SOL野生型纤维中为1.2±0.4×10⁻⁶ M,但在Kir6.1纤维中无法测量。Kir6.1纤维中KATP通道对MgATP的敏感性未改变。对SOL的组织病理学/免疫组织化学分析显示,在Kir6.1小鼠中存在变性以及退行性坏死性病变并伴有再生,且Atrogin-1、MuRF1和BNIP3的mRNA/蛋白上调。骨骼肌中的Kir6.1突变导致FDB和SOL纤维中KATP通道对格列本脲的反应发生变化,并与慢肌中的组织病理学和基因表达变化相关,提示明显萎缩和自噬。