Gardina Paul J, Lo Ken C, Lee Walter, Cowell John K, Turpaz Yaron
Affymetrix, Inc., 3420 Central Expressway, Santa Clara, California 95051, USA.
BMC Genomics. 2008 Oct 17;9:489. doi: 10.1186/1471-2164-9-489.
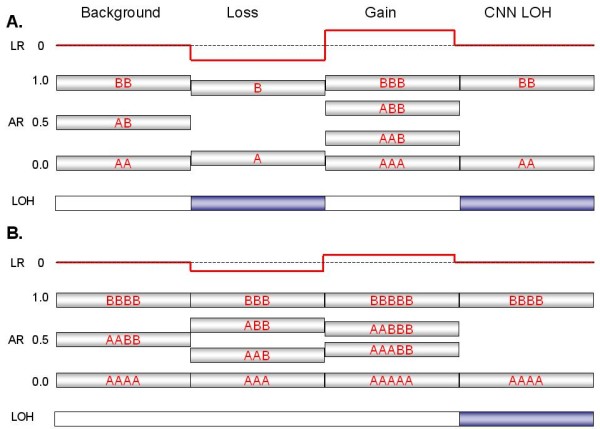
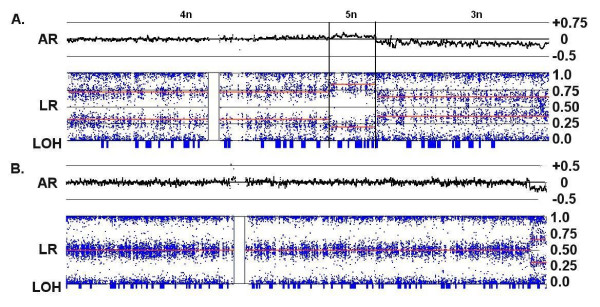
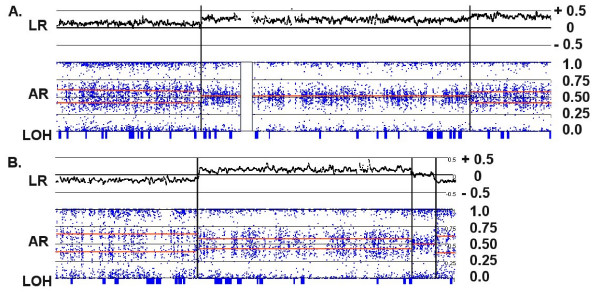
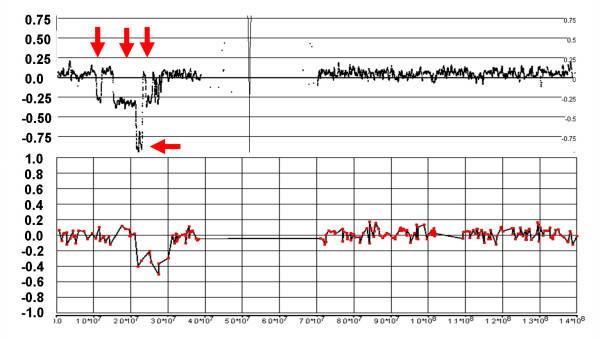
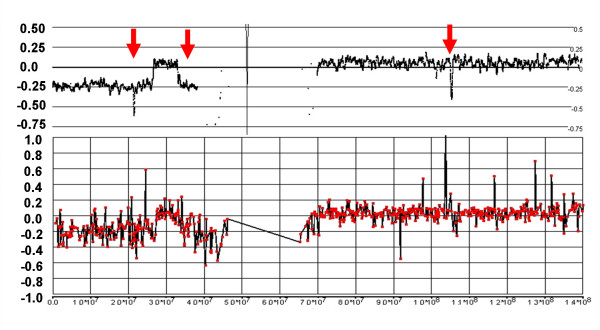
Genomic hybridization platforms, including BAC-CGH and genotyping arrays, have been used to estimate chromosome copy number (CN) in tumor samples by detecting the relative strength of genomic signal. The methods rely on the assumption that the predominant chromosomal background of the samples is diploid, an assumption that is frequently incorrect for tumor samples. In addition to generally greater resolution, an advantage of genotyping arrays over CGH arrays is the ability to detect signals from individual alleles, allowing estimation of loss-of-heterozygosity (LOH) and allelic ratios to enhance the interpretation of copy number alterations. Copy number events associated with LOH potentially have the same genetic consequences as deletions.
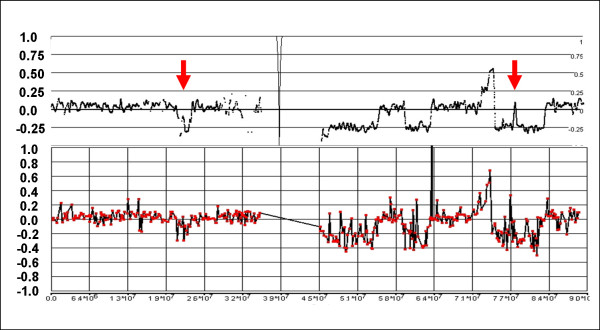
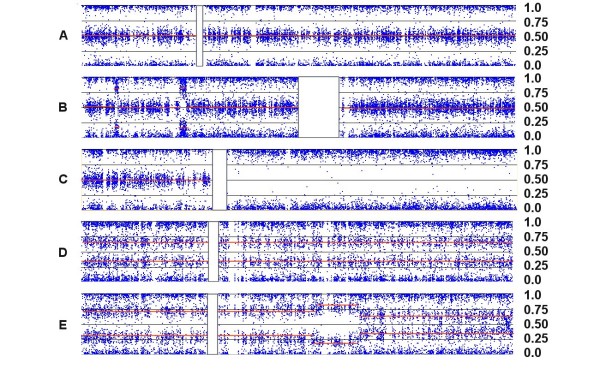
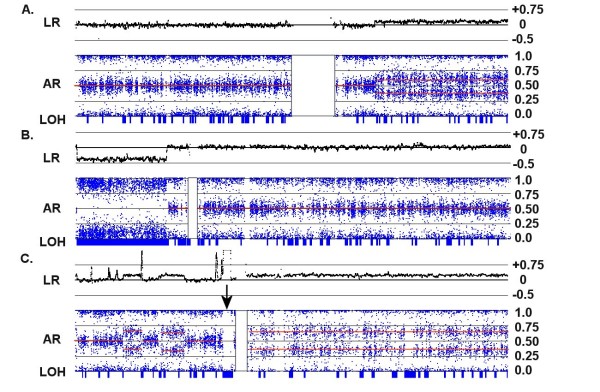
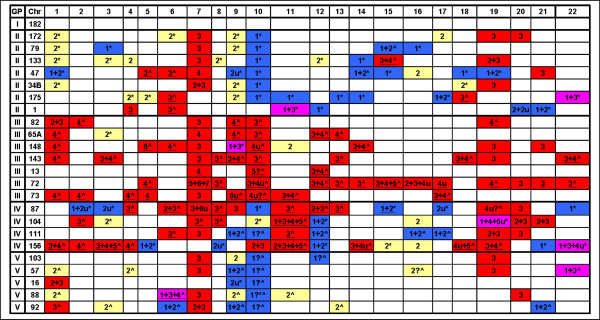
We have utilized allelic ratios to detect patterns that are indicative of higher ploidy levels. An integrated analysis using allelic ratios, total signal and LOH indicates that many or most of the chromosomes from 24 glioblastoma tumors are in fact aneuploid. Some putative whole-chromosome losses actually represent trisomy, and many apparent sub-chromosomal losses are in fact relative losses against a triploid or tetraploid background.
These results suggest a re-interpretation of previous findings based only on total signal ratios. One interesting observation is that many single or multiple-copy deletions occur at common putative tumor suppressor sites subsequent to chromosomal duplication; these losses do not necessarily result in LOH, but nonetheless occur in conspicuous patterns. The 500 K Mapping array was also capable of detecting many sub-mega base losses and gains that were overlooked by CGH-BAC arrays, and was superior to CGH-BAC arrays in resolving regions of complex CN variation.
基因组杂交平台,包括细菌人工染色体比较基因组杂交(BAC-CGH)和基因分型阵列,已被用于通过检测基因组信号的相对强度来估计肿瘤样本中的染色体拷贝数(CN)。这些方法依赖于样本的主要染色体背景是二倍体这一假设,而这一假设对于肿瘤样本来说常常是不正确的。除了通常具有更高的分辨率外,基因分型阵列相对于CGH阵列的一个优势是能够检测来自单个等位基因的信号,从而能够估计杂合性缺失(LOH)和等位基因比例,以增强对拷贝数改变的解释。与LOH相关的拷贝数事件可能与缺失具有相同的遗传后果。
我们利用等位基因比例来检测指示更高倍性水平的模式。使用等位基因比例、总信号和LOH的综合分析表明,24例胶质母细胞瘤肿瘤中的许多或大多数染色体实际上是非整倍体。一些假定的全染色体缺失实际上代表三体性,许多明显的亚染色体缺失实际上是相对于三倍体或四倍体背景的相对缺失。
这些结果表明需要对仅基于总信号比例的先前发现进行重新解释。一个有趣的观察结果是,许多单拷贝或多拷贝缺失发生在染色体复制后的常见假定肿瘤抑制位点;这些缺失不一定导致LOH,但仍然以明显的模式出现。500K Mapping阵列还能够检测到许多被CGH-BAC阵列忽略的亚兆碱基缺失和增益,并且在解析复杂CN变异区域方面优于CGH-BAC阵列。