Unité de Recherche en Biologie Moléculaire, Facultés Universitaires Notre-Dame de la Paix (F.U.N.D.P.), Rue de Bruxelles 61, B-5000 Namur, Belgium.
BMC Bioinformatics. 2010 Jan 11;11:17. doi: 10.1186/1471-2105-11-17.
Recent reanalysis of spike-in datasets underscored the need for new and more accurate benchmark datasets for statistical microarray analysis. We present here a fresh method using biologically-relevant data to evaluate the performance of statistical methods.
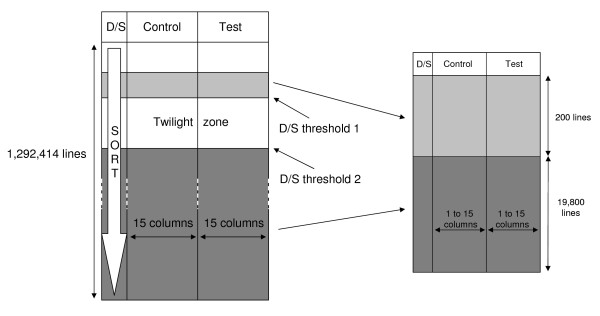
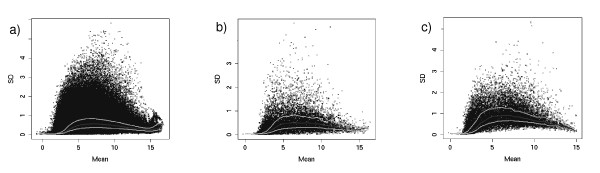
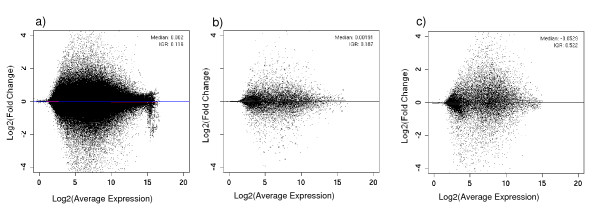
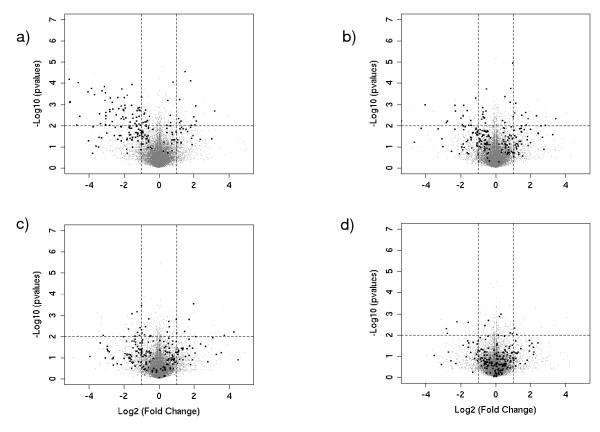
Our novel method ranks the probesets from a dataset composed of publicly-available biological microarray data and extracts subset matrices with precise information/noise ratios. Our method can be used to determine the capability of different methods to better estimate variance for a given number of replicates. The mean-variance and mean-fold change relationships of the matrices revealed a closer approximation of biological reality.
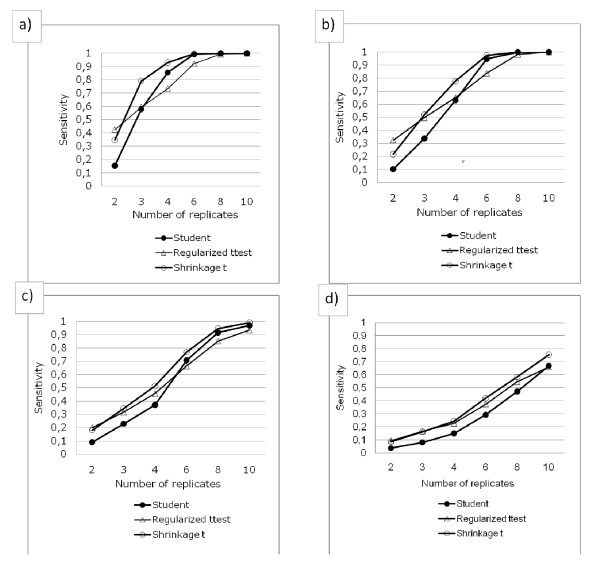
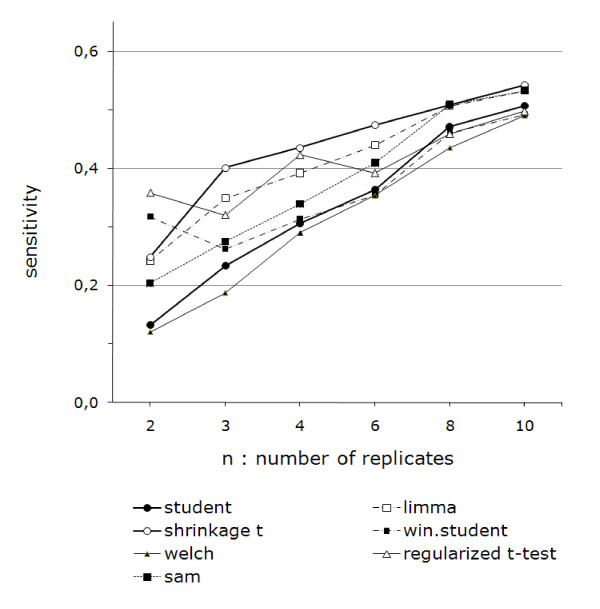
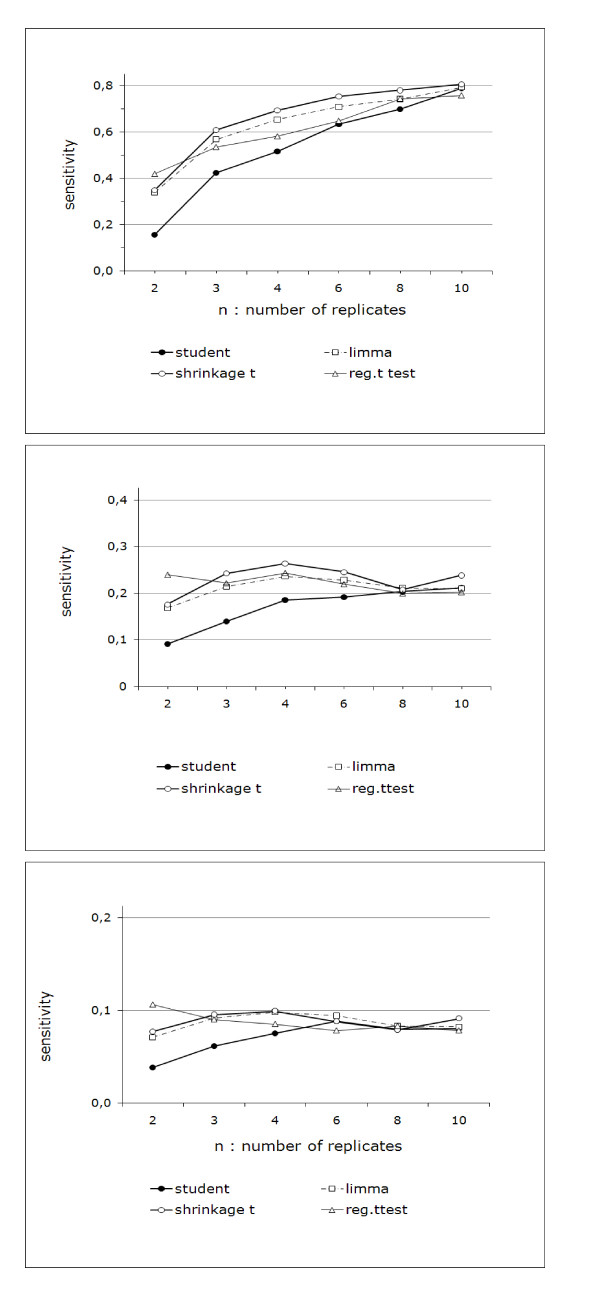
Performance analysis refined the results from benchmarks published previously.We show that the Shrinkage t test (close to Limma) was the best of the methods tested, except when two replicates were examined, where the Regularized t test and the Window t test performed slightly better.
The R scripts used for the analysis are available at http://urbm-cluster.urbm.fundp.ac.be/~bdemeulder/.
最近对 Spike-in 数据集的重新分析强调了需要新的、更准确的统计微阵列分析基准数据集。我们在这里提出了一种使用生物学相关数据来评估统计方法性能的新方法。
我们的新方法对由公开可用的生物学微阵列数据组成的数据集进行了探针集排序,并提取了具有精确信息/噪声比的子矩阵。我们的方法可用于确定不同方法在给定重复次数下更好地估计方差的能力。矩阵的均值方差和均值倍数变化关系更接近生物学实际情况。
性能分析改进了先前发布的基准测试的结果。我们表明,收缩 t 检验(接近 Limma)是测试的方法中最好的方法,除了当检查两个重复时,正则化 t 检验和窗口 t 检验的性能略好一些。
用于分析的 R 脚本可在 http://urbm-cluster.urbm.fundp.ac.be/~bdemeulder/ 获得。