Department of Medical Genetics, University of Medical Sciences, ul. Grunwaldzka 55 paw. 15, 60-352 Poznań, Poland.
J Appl Genet. 2012 Nov;53(4):415-22. doi: 10.1007/s13353-012-0109-x. Epub 2012 Aug 18.
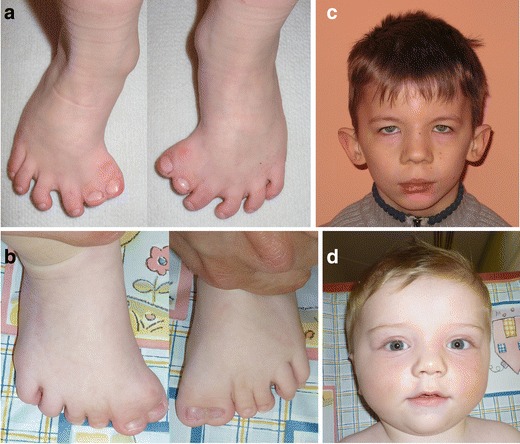
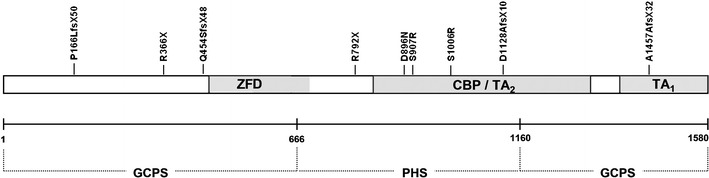
Greig cephalopolysyndactyly syndrome (GCPS) and isolated preaxial polydactyly type IV (PPD-IV) are rare autosomal dominant disorders, both caused by mutations in the GLI3 gene. GCPS is mainly characterised by craniofacial abnormalities (macrocephaly/prominent forehead, hypertelorism) and limb malformations, such as PPD-IV, syndactyly and postaxial polydactyly type A or B (PAPA/B). Mutations in the GLI3 gene can also lead to Pallister-Hall syndrome (PHS) and isolated PAPA/B. In this study, we investigated 16 unrelated probands with the clinical diagnosis of GCPS/PPD-IV and found GLI3 mutations in 12 (75%) of them (nine familial and three sporadic cases). We also performed a detailed clinical evaluation of all 12 GLI3-positive families, with a total of 27 patients. The hallmark triad of GCPS (preaxial polydactyly, macrocephaly/prominent forehead, hypertelorism) was present in 14 cases (52%), whereas at least one typical dysmorphic feature was manifested in 17 patients (63%). Upon sequencing of the GLI3 gene, we demonstrated eight novel and two previously reported heterozygous point mutations. We also performed multiplex ligation-dependent probe amplification (MLPA) to screen for intragenic copy number changes and identified heterozygous deletions in the two remaining cases (16.7%). Our findings fully support previous genotype-phenotype correlations, showing that exonic deletions, missense mutations, as well as truncating variants localised out of the middle third of the GLI3 gene result in GCPS/PPD-IV and not PHS. Additionally, our study shows that intragenic GLI3 deletions may account for a significant proportion of GCPS/PPD-IV causative mutations. Therefore, we propose that MLPA or quantitative polymerase chain reaction (qPCR) should be implemented into routine molecular diagnostic of the GLI3 gene.
格里希颅面多并指综合征(GCPS)和孤立性桡侧多指畸形 IV 型(PPD-IV)是罕见的常染色体显性遗传疾病,均由 GLI3 基因突变引起。GCPS 的主要特征是颅面异常(大头畸形/额突出、眼距过宽)和肢体畸形,如 PPD-IV、并指和轴后多指 A 或 B 型(PAPA/B)。GLI3 基因突变也可导致帕利斯特-霍尔综合征(PHS)和孤立性 PAPA/B。在这项研究中,我们对 16 名临床诊断为 GCPS/PPD-IV 的无关先证者进行了研究,发现 12 名(75%)患者存在 GLI3 基因突变(9 个家系性病例和 3 个散发性病例)。我们还对所有 12 个 GLI3 阳性家系进行了详细的临床评估,共涉及 27 名患者。14 例(52%)存在 GCPS 的三联征(桡侧多指、大头畸形/额突出、眼距过宽),17 例(63%)至少表现出一个典型的畸形特征。在对 GLI3 基因进行测序后,我们发现了 8 个新的和 2 个以前报道的杂合点突变。我们还进行了多重连接依赖性探针扩增(MLPA)来筛选基因内拷贝数变化,并在另外 2 个病例中发现了杂合性缺失(16.7%)。我们的发现完全支持以前的基因型-表型相关性,表明外显子缺失、错义突变以及位于 GLI3 基因中段以外的截断变异导致 GCPS/PPD-IV,而不是 PHS。此外,我们的研究表明,基因内 GLI3 缺失可能占 GCPS/PPD-IV 致病突变的很大比例。因此,我们建议将 MLPA 或实时定量聚合酶链反应(qPCR)纳入 GLI3 基因的常规分子诊断中。