NIH Undiagnosed Diseases Program, Common Fund, Office of the Director, NIH and Office of the Clinical Director, National Human Genome Research Institute, NIH, Bethesda, 20892, Maryland, USA.
Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, 20892, Maryland, USA.
Hum Mutat. 2018 Jan;39(1):69-79. doi: 10.1002/humu.23345. Epub 2017 Nov 8.
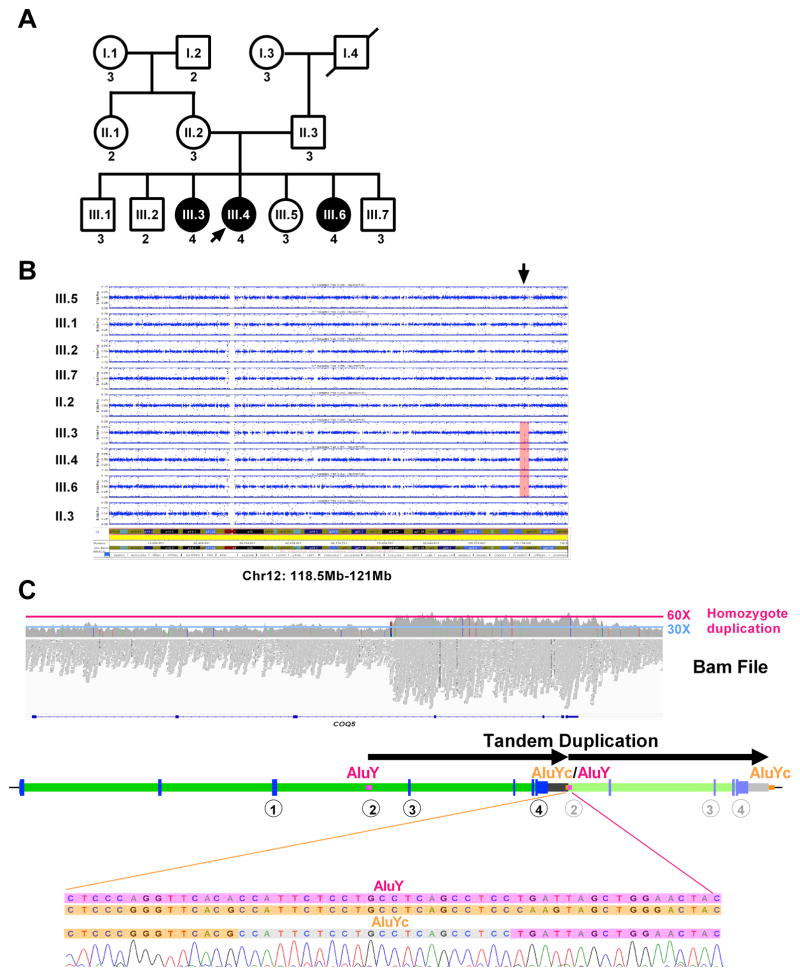
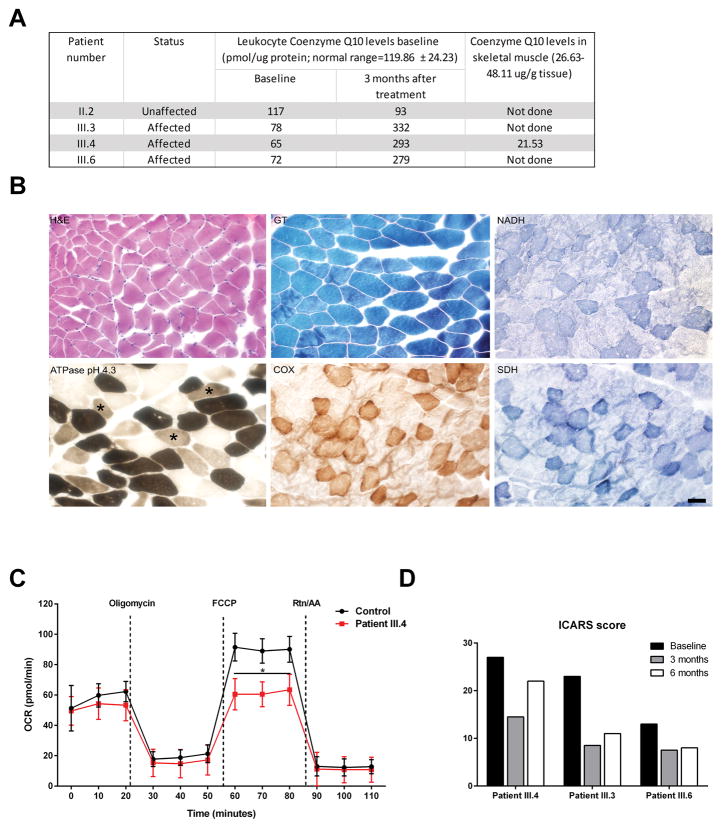
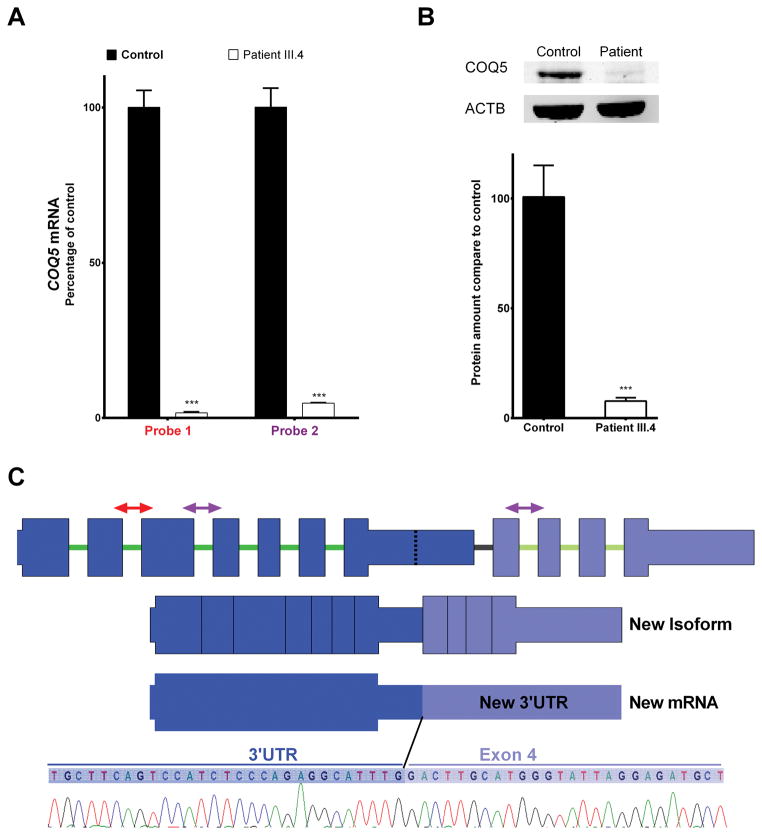
Primary coenzyme Q10 (CoQ ; MIM# 607426) deficiencies are an emerging group of inherited mitochondrial disorders with heterogonous clinical phenotypes. Over a dozen genes are involved in the biosynthesis of CoQ , and mutations in several of these are associated with human disease. However, mutations in COQ5 (MIM# 616359), catalyzing the only C-methylation in the CoQ synthetic pathway, have not been implicated in human disease. Here, we report three female siblings of Iraqi-Jewish descent, who had varying degrees of cerebellar ataxia, encephalopathy, generalized tonic-clonic seizures, and cognitive disability. Whole-exome and subsequent whole-genome sequencing identified biallelic duplications in the COQ5 gene, leading to reduced levels of CoQ in peripheral white blood cells of all affected individuals and reduced CoQ levels in the only muscle tissue available from one affected proband. CoQ supplementation led to clinical improvement and increased the concentrations of CoQ in blood. This is the first report of primary CoQ deficiency caused by loss of function of COQ5, with delineation of the clinical, laboratory, histological, and molecular features, and insights regarding targeted treatment with CoQ supplementation.
原发性辅酶 Q10(CoQ;MIM#607426)缺乏症是一组新兴的遗传性线粒体疾病,具有异质的临床表型。辅酶 Q 生物合成涉及十多个基因,其中一些基因的突变与人类疾病有关。然而,催化辅酶 Q 合成途径中唯一的 C-甲基化的 COQ5(MIM#616359)基因突变尚未与人类疾病相关。在这里,我们报告了三个具有伊拉克-犹太血统的女性兄弟姐妹,她们具有不同程度的小脑共济失调、脑病、全身性强直阵挛性癫痫发作和认知障碍。全外显子组和随后的全基因组测序确定了 COQ5 基因的双等位基因重复,导致所有受影响个体的外周白细胞中 CoQ 水平降低,并且仅从一名受影响的先证者获得的肌肉组织中 CoQ 水平降低。CoQ 补充剂治疗导致了临床改善,并增加了血液中的 CoQ 浓度。这是首次报道由 COQ5 功能丧失引起的原发性 CoQ 缺乏症,阐述了其临床、实验室、组织学和分子特征,以及针对 CoQ 补充剂的靶向治疗的见解。