Life Sciences Institute and Innovation Center for Cell Signaling Network, Zhejiang University, Hangzhou, Zhejiang, 310058, China.
School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pok Fu Lam, Hong Kong, China.
Nat Commun. 2017 Nov 13;8(1):1470. doi: 10.1038/s41467-017-01759-y.
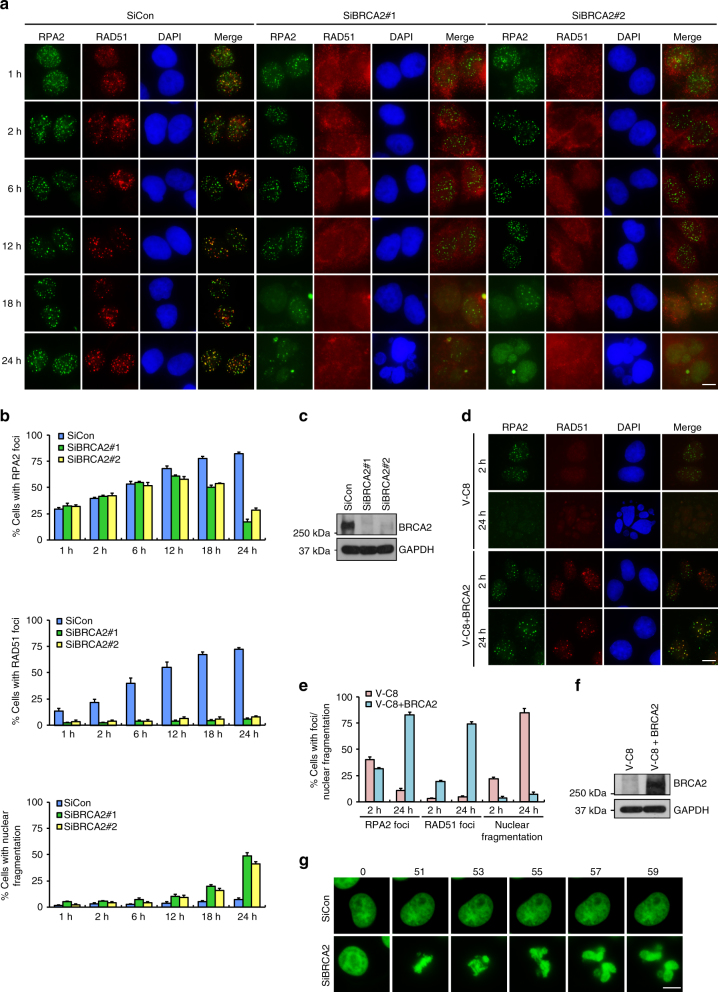
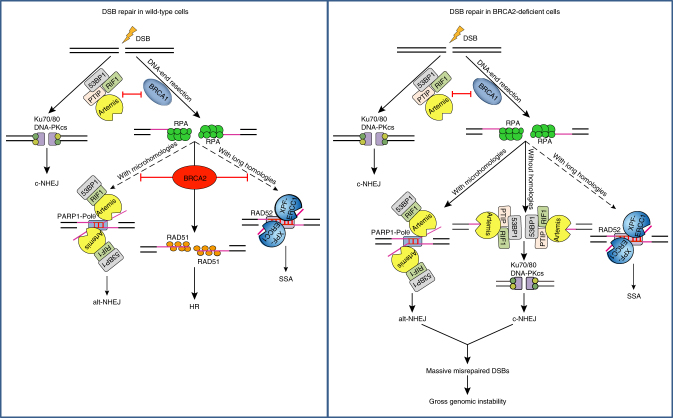
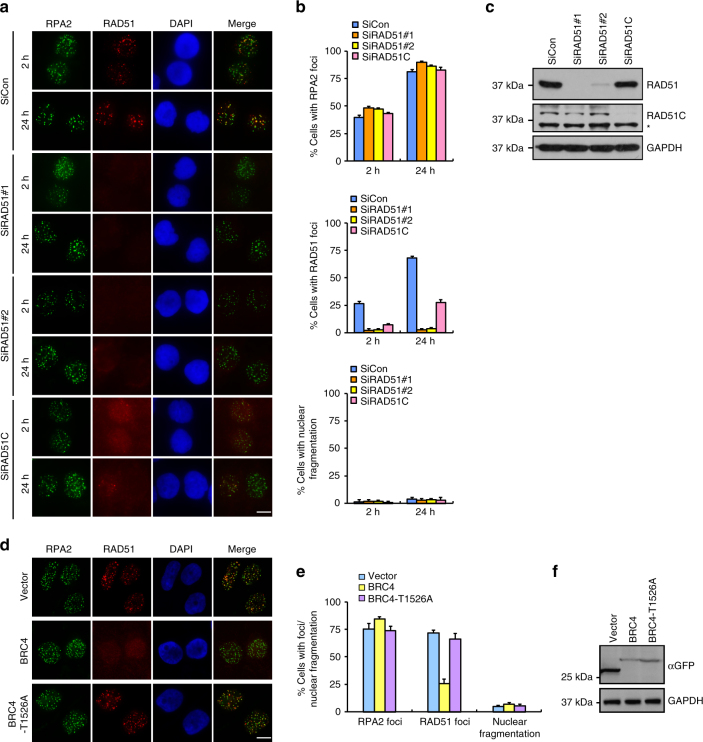
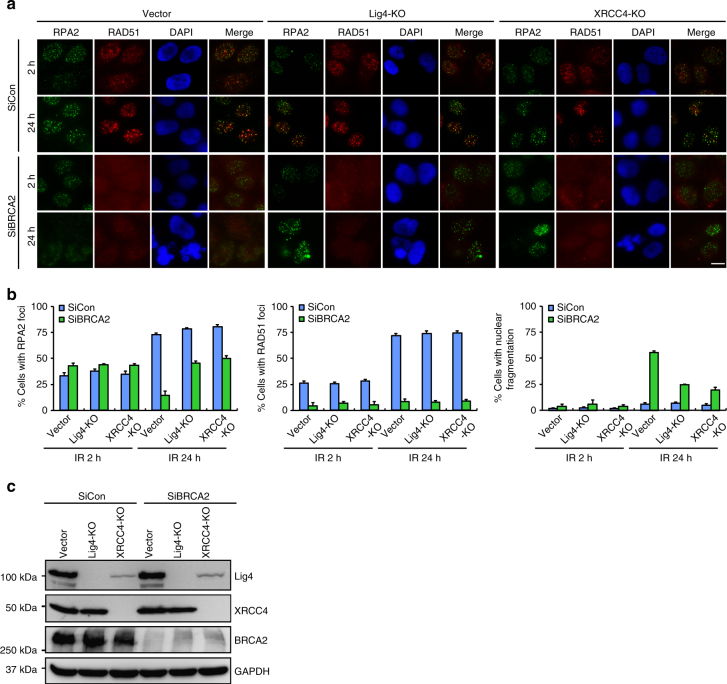
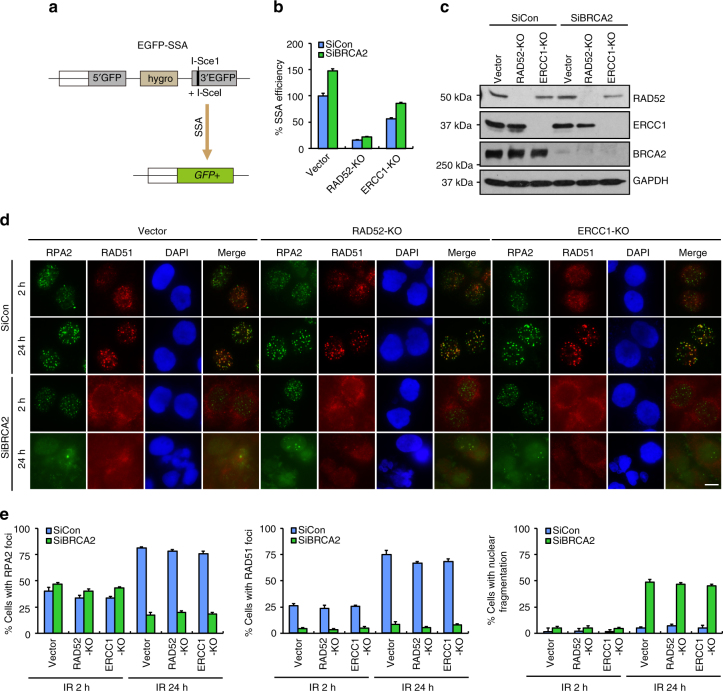
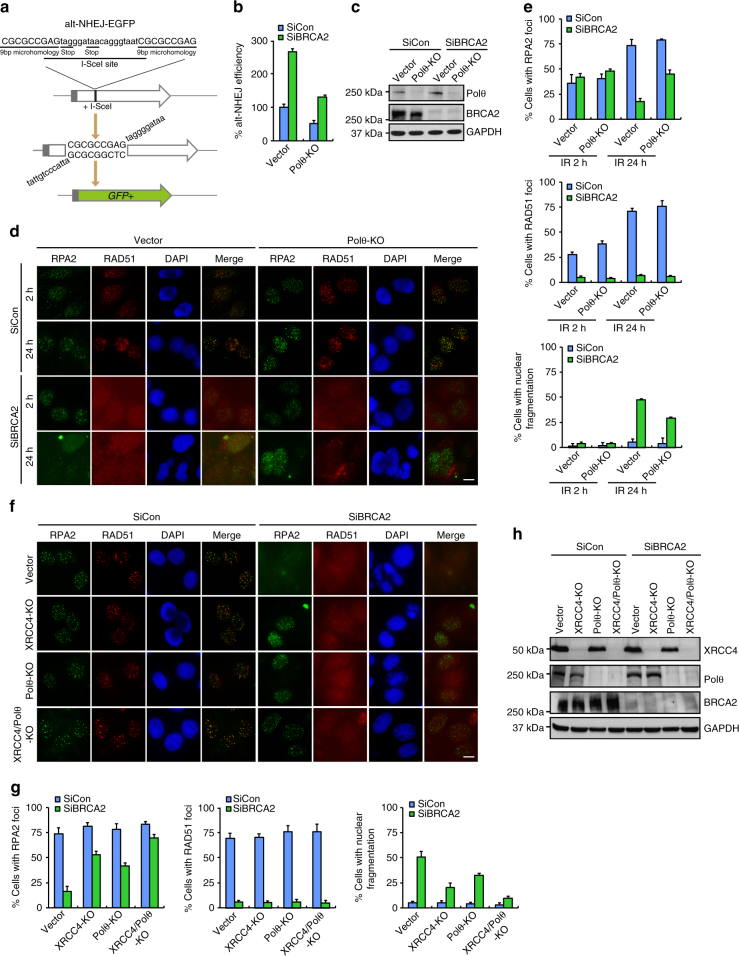
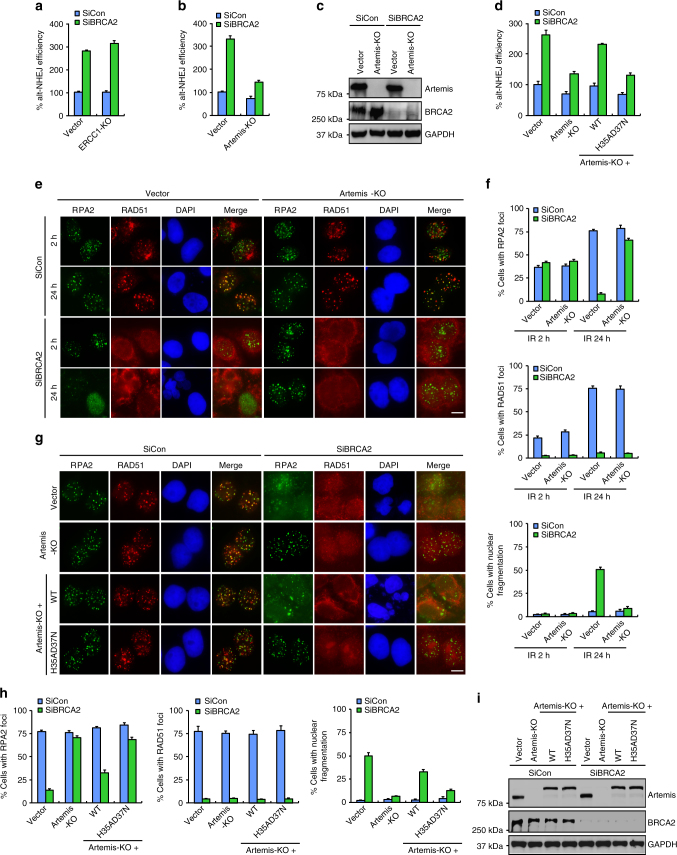
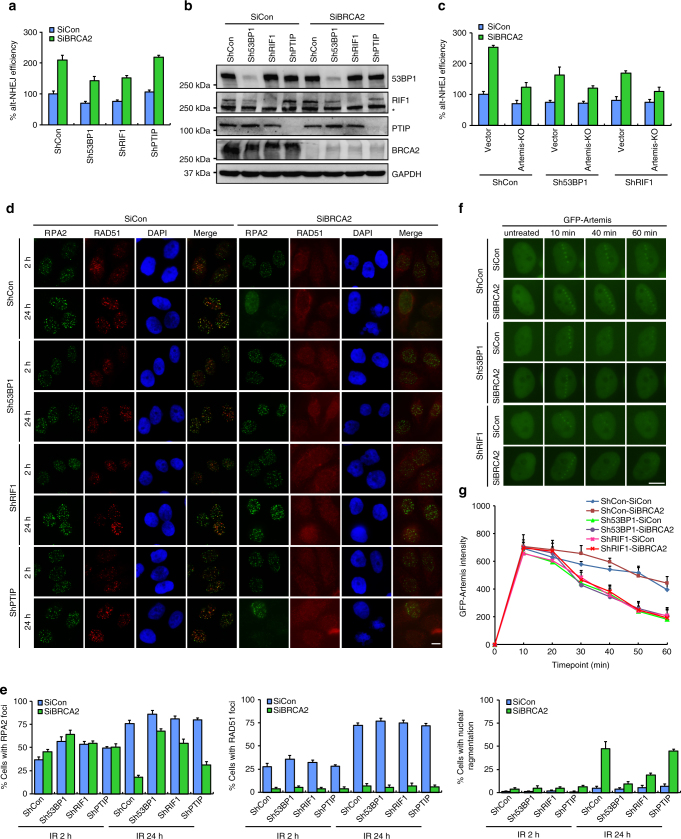
BRCA2-deficient cells exhibit gross genomic instability, but the underlying mechanisms are not fully understood. Here we report that inactivation of BRCA2 but not RAD51 destabilizes RPA-coated single-stranded DNA (ssDNA) structures at resected DNA double-strand breaks (DSBs) and greatly enhances the frequency of nuclear fragmentation following cell exposure to DNA damage. Importantly, these BRCA2-associated deficits are fueled by the aberrant activation of classical (c)- and alternative (alt)- nonhomologous end-joining (NHEJ), and rely on the well-defined DNA damage signaling pathway involving the pro-c-NHEJ factor 53BP1 and its downstream effector RIF1. We further show that the 53BP1-RIF1 axis promotes toxic end-joining events via the retention of Artemis at DNA damage sites. Accordingly, loss of 53BP1, RIF1, or Artemis prolongs the stability of RPA-coated DSB intermediates in BRCA2-deficient cells and restores nuclear integrity. We propose that BRCA2 antagonizes 53BP1, RIF1, and Artemis-dependent c-NHEJ and alt-NHEJ to prevent gross genomic instability in a RAD51-independent manner.
BRCA2 缺陷细胞表现出明显的基因组不稳定性,但潜在的机制尚不完全清楚。在这里,我们报告 BRCA2 的失活而不是 RAD51 的失活会破坏在切除的 DNA 双链断裂(DSB)处的 RPA 包裹的单链 DNA(ssDNA)结构,并在细胞暴露于 DNA 损伤后大大增加核碎片的频率。重要的是,这些与 BRCA2 相关的缺陷是由经典(c)和替代(alt)非同源末端连接(NHEJ)的异常激活所驱动的,并且依赖于涉及 pro-c-NHEJ 因子 53BP1 及其下游效应子 RIF1 的明确的 DNA 损伤信号通路。我们进一步表明,53BP1-RIF1 轴通过在 DNA 损伤部位保留 Artemis 来促进有毒的末端连接事件。因此,53BP1、RIF1 或 Artemis 的缺失会延长 BRCA2 缺陷细胞中 RPA 包裹的 DSB 中间体的稳定性,并恢复核完整性。我们提出 BRCA2 以 RAD51 独立的方式拮抗 53BP1、RIF1 和 Artemis 依赖性 c-NHEJ 和 alt-NHEJ,以防止明显的基因组不稳定性。