Department of Electrical Engineering and Computer Sciences, University of California, Berkeley, Berkeley, United States.
Department of Statistics, University of California, Berkeley, Berkeley, United States.
Elife. 2018 Jun 13;7:e32920. doi: 10.7554/eLife.32920.
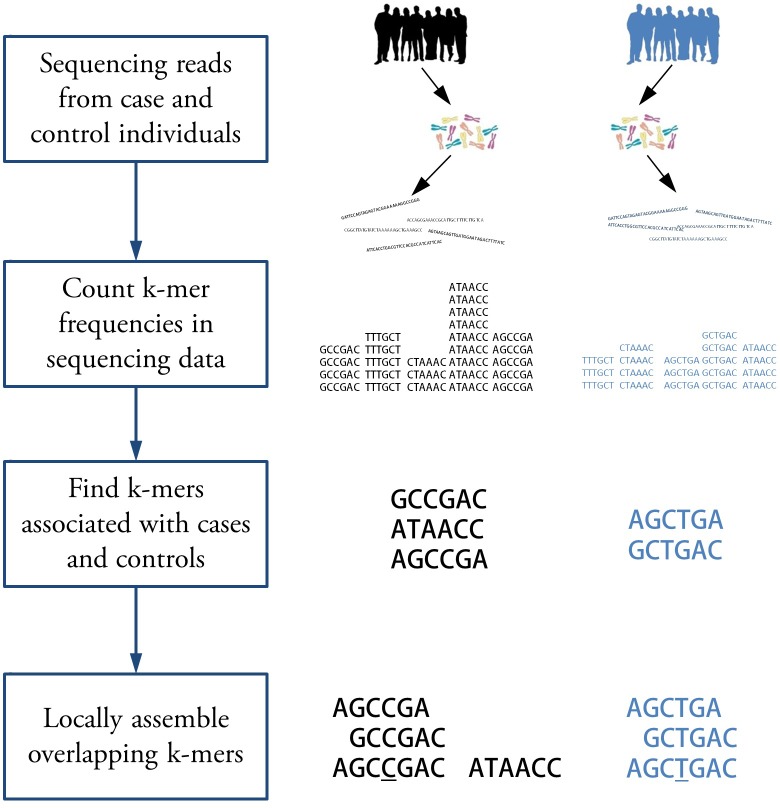
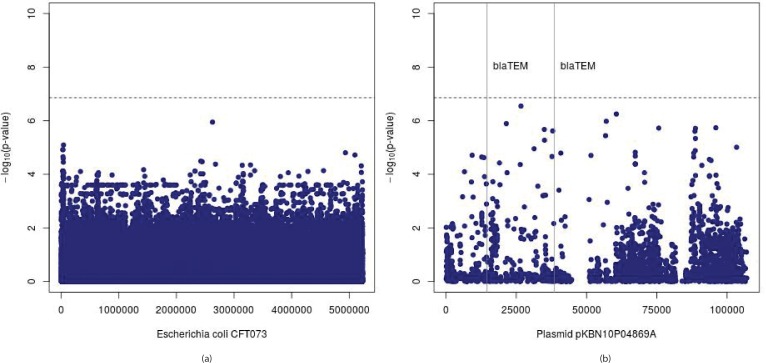
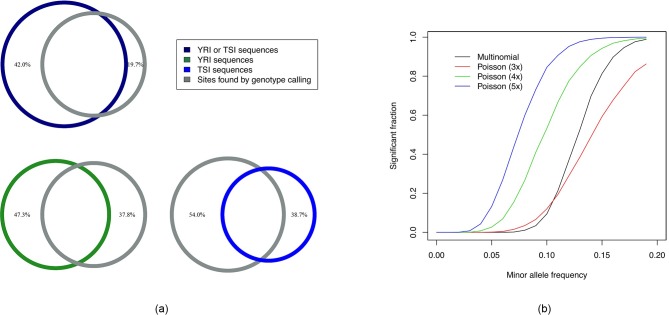
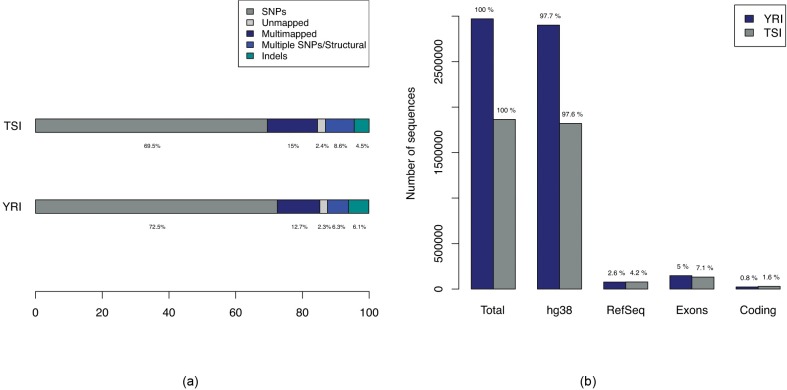
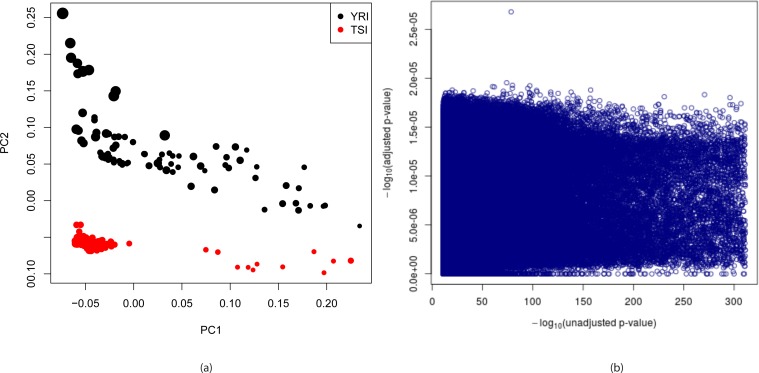
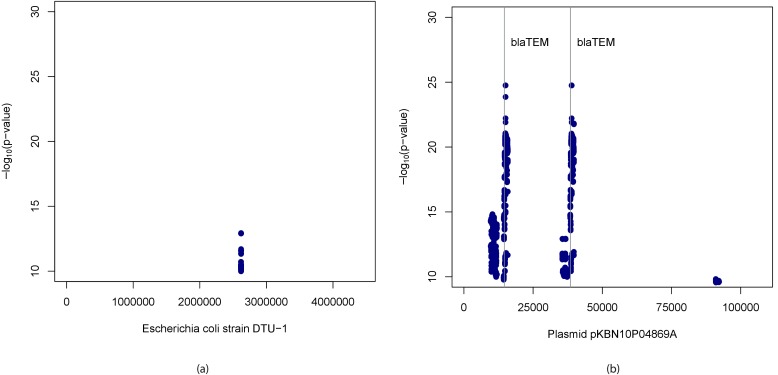
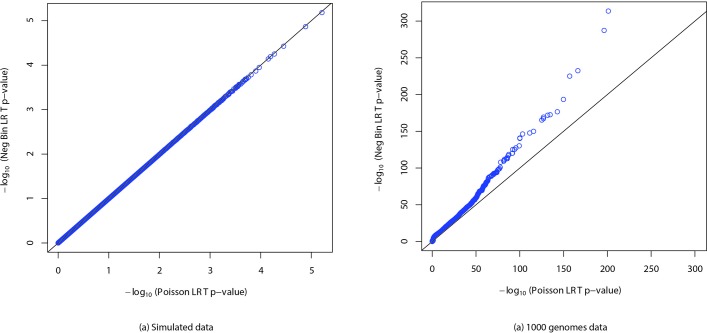
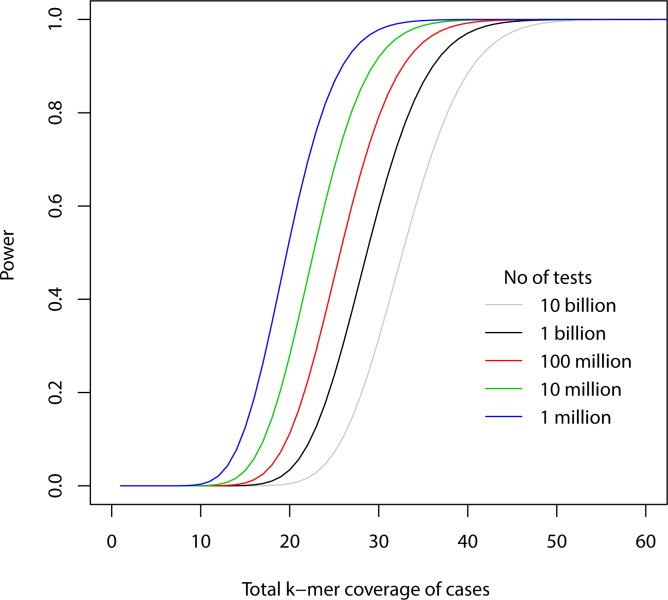
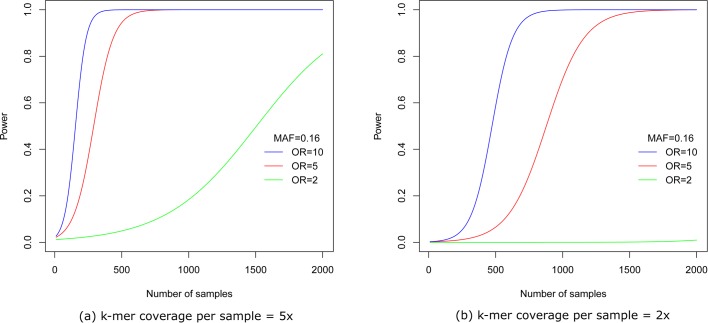
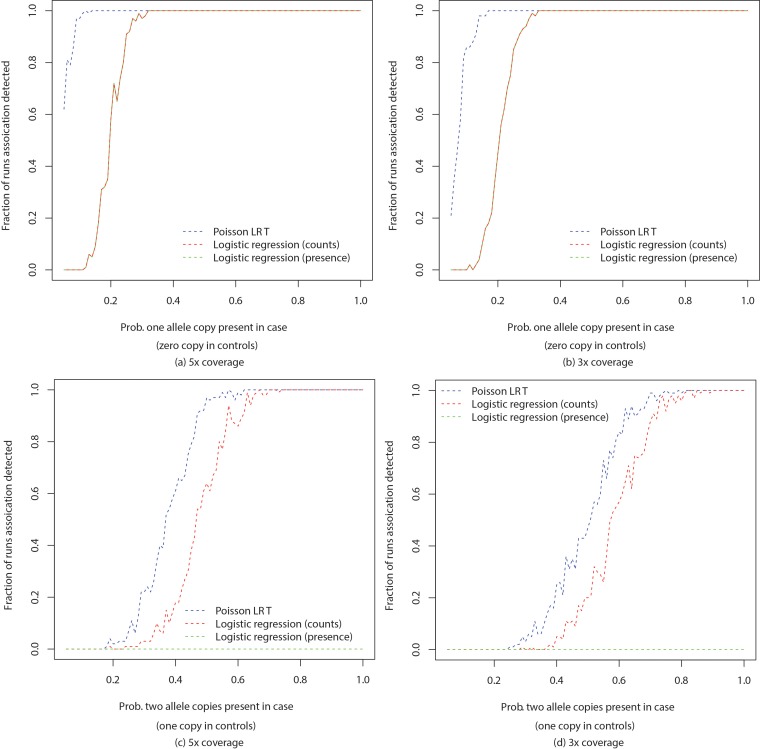
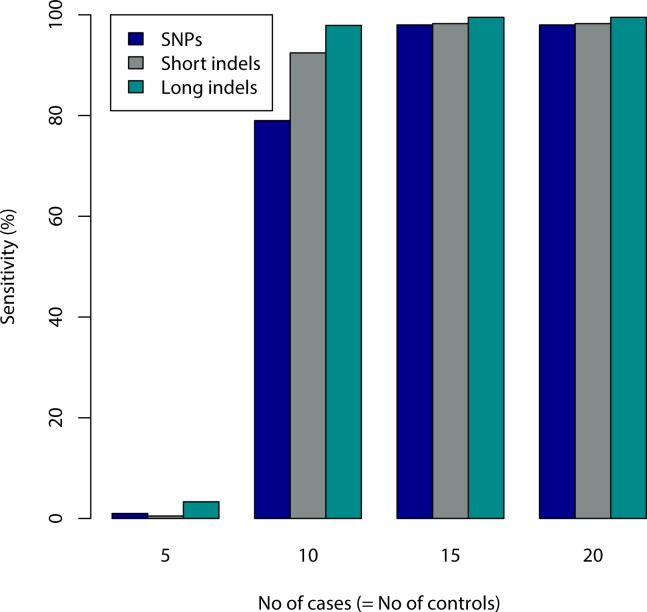
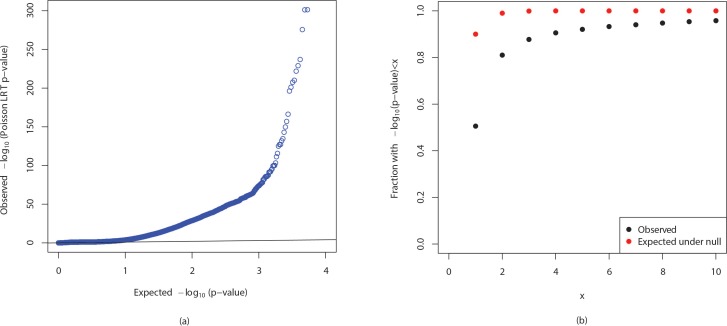
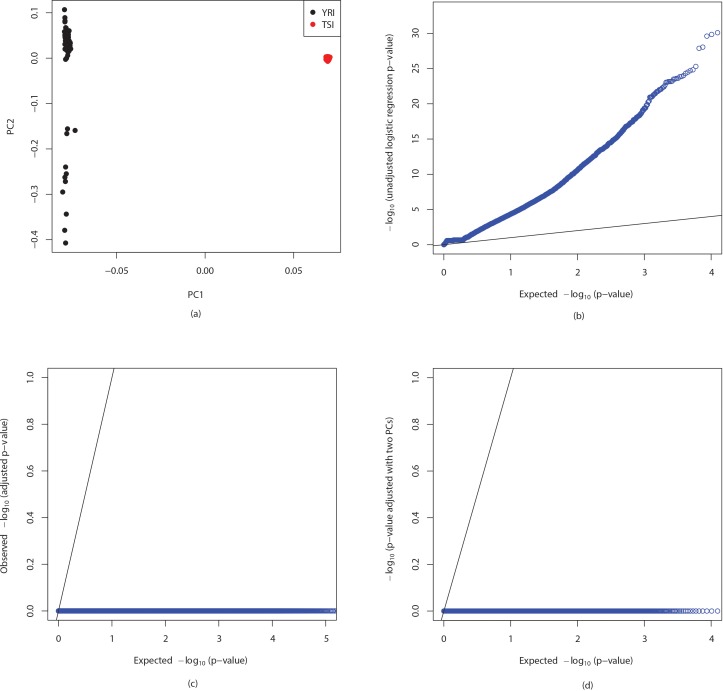
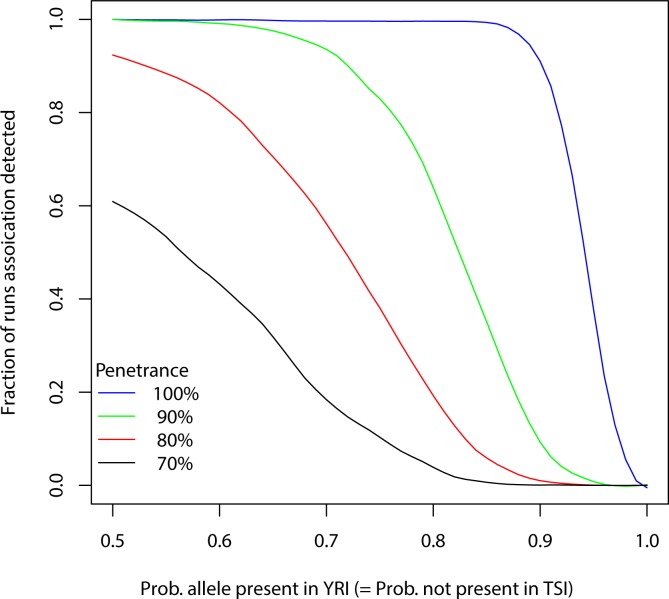
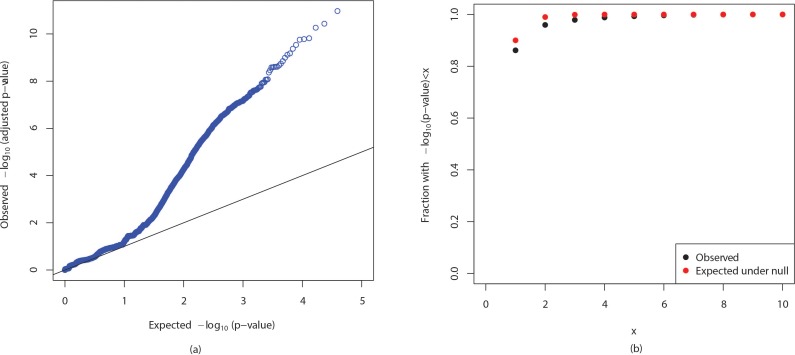
Genome wide association studies (GWAS) rely on microarrays, or more recently mapping of sequencing reads, to genotype individuals. The reliance on prior sequencing of a reference genome limits the scope of association studies, and also precludes mapping associations outside of the reference. We present an alignment free method for association studies of categorical phenotypes based on counting [Formula: see text]-mers in whole-genome sequencing reads, testing for associations directly between [Formula: see text]-mers and the trait of interest, and local assembly of the statistically significant [Formula: see text]-mers to identify sequence differences. An analysis of the 1000 genomes data show that sequences identified by our method largely agree with results obtained using the standard approach. However, unlike standard GWAS, our method identifies associations with structural variations and sites not present in the reference genome. We also demonstrate that population stratification can be inferred from [Formula: see text]-mers. Finally, application to an dataset on ampicillin resistance validates the approach.
全基因组关联研究(GWAS)依赖于微阵列,或最近的测序读段映射,对个体进行基因分型。对参考基因组进行先前测序的依赖限制了关联研究的范围,并且也排除了参考之外的关联映射。我们提出了一种基于全基因组测序读段中计数 [Formula: see text]-mers 的分类表型关联研究的无比对方法,直接在 [Formula: see text]-mers 和感兴趣的性状之间进行关联测试,并对统计学上显著的 [Formula: see text]-mers 进行局部组装,以识别序列差异。对 1000 基因组数据的分析表明,我们的方法识别的序列在很大程度上与使用标准方法获得的结果一致。然而,与标准的 GWAS 不同,我们的方法可以识别与结构变异和参考基因组中不存在的位点相关的关联。我们还证明了可以从 [Formula: see text]-mers 推断出种群分层。最后,对氨苄青霉素抗性数据集的应用验证了该方法。