Department of Information Engineering, University of Padova, via Gradenigo 6/A, Padova, Italy.
BMC Bioinformatics. 2018 Nov 30;19(Suppl 15):441. doi: 10.1186/s12859-018-2415-8.
Spaced-seeds, i.e. patterns in which some fixed positions are allowed to be wild-cards, play a crucial role in several bioinformatics applications involving substrings counting and indexing, by often providing better sensitivity with respect to k-mers based approaches. K-mers based approaches are usually fast, being based on efficient hashing and indexing that exploits the large overlap between consecutive k-mers. Spaced-seeds hashing is not as straightforward, and it is usually computed from scratch for each position in the input sequence. Recently, the FSH (Fast Spaced seed Hashing) approach was proposed to improve the time required for computation of the spaced seed hashing of DNA sequences with a speed-up of about 1.5 with respect to standard hashing computation.
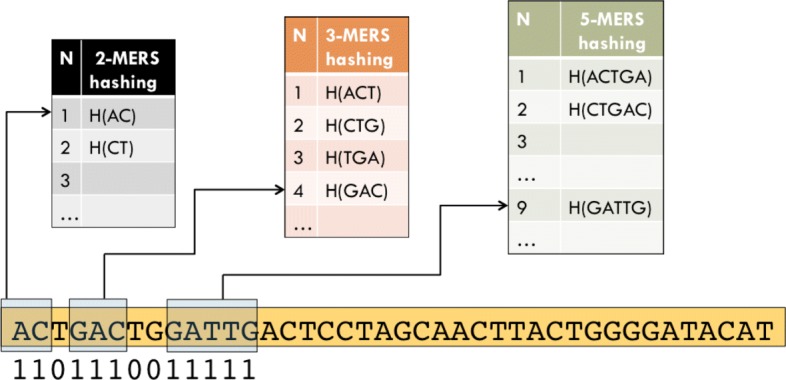
In this work we propose a novel algorithm, Fast Indexing for Spaced seed Hashing (FISH), based on the indexing of small blocks that can be combined to obtain the hashing of spaced-seeds of any length. The method exploits the fast computation of the hashing of runs of consecutive 1 in the spaced seeds, that basically correspond to k-mer of the length of the run.
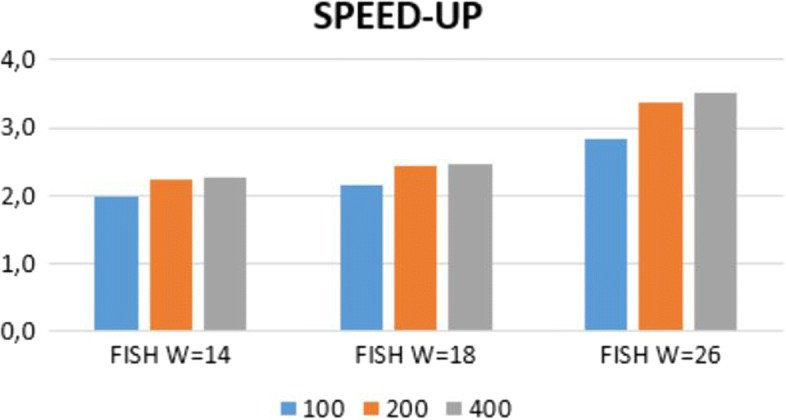
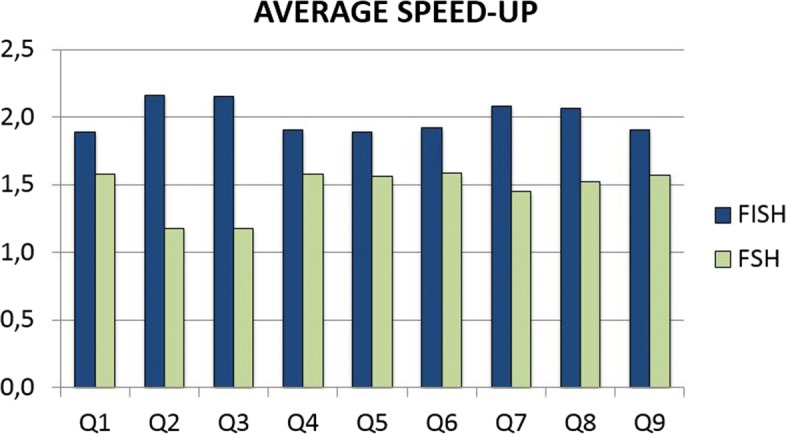
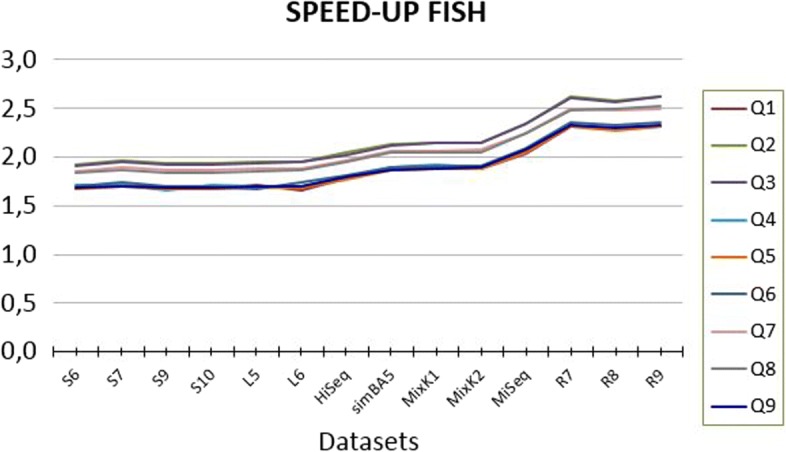
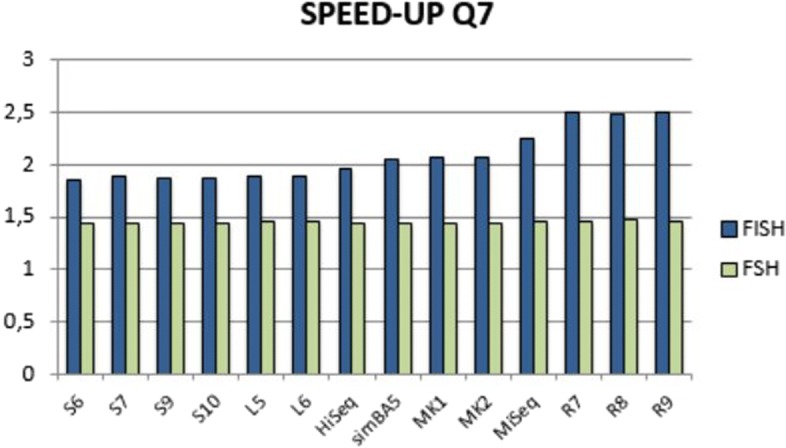
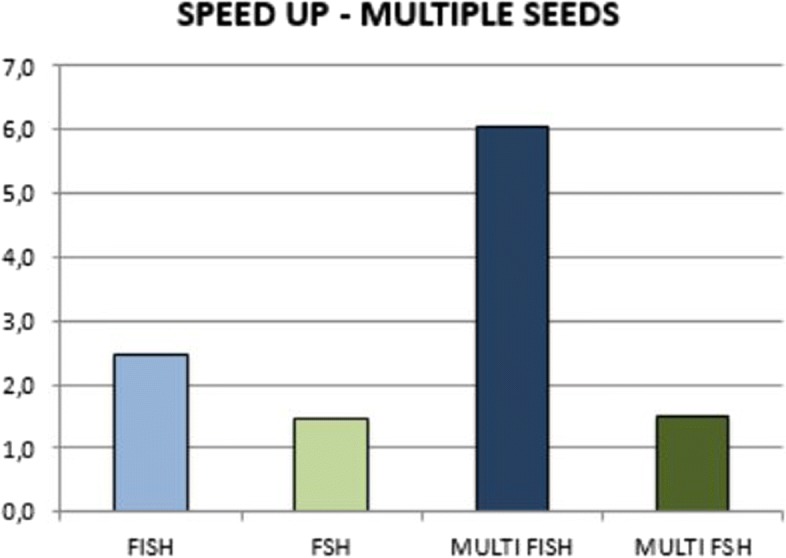
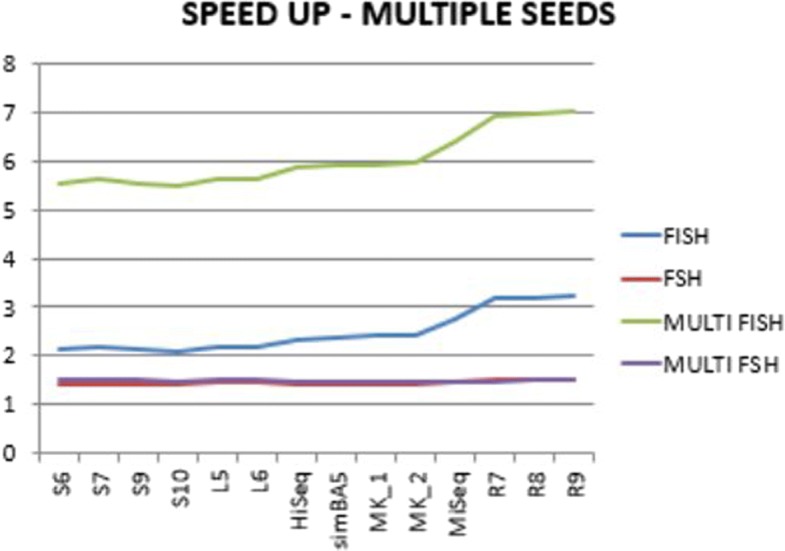
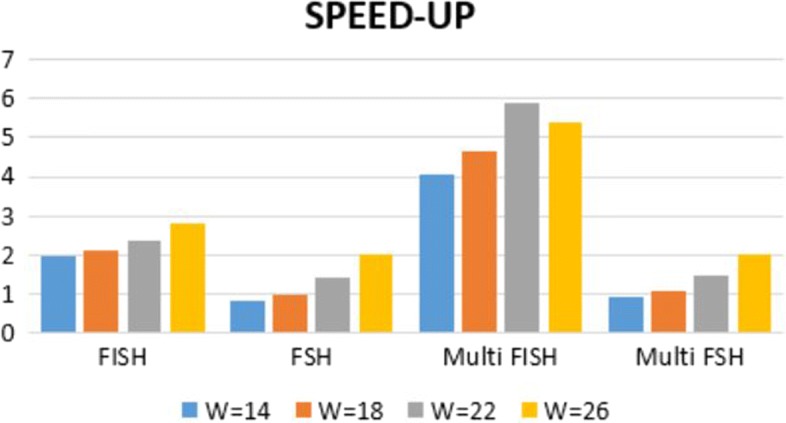
We run several experiments, on NGS data from simulated and synthetic metagenomic experiments, to assess the time required for the computation of the hashing for each position in each read with respect to several spaced seeds. In our experiments, FISH can compute the hashing values of spaced seeds with a speedup, with respect to the traditional approach, between 1.9x to 6.03x, depending on the structure of the spaced seeds.
间隔种子(即某些固定位置允许使用通配符的模式)在涉及子串计数和索引的几个生物信息学应用中起着至关重要的作用,通常相对于基于 k-mer 的方法提供更好的敏感性。基于 k-mer 的方法通常很快,因为它们基于利用连续 k-mer 之间的大量重叠的高效哈希和索引。间隔种子哈希并不那么直接,通常需要为输入序列中的每个位置从头开始计算。最近,提出了 FSH(Fast Spaced seed Hashing)方法来提高计算 DNA 序列间隔种子哈希的时间,相对于标准哈希计算,速度提高了约 1.5 倍。
在这项工作中,我们提出了一种新的算法,即间隔种子哈希的快速索引(Fast Indexing for Spaced seed Hashing,FISH),该算法基于可以组合以获得任意长度间隔种子哈希的小块索引。该方法利用间隔种子中连续 1 位的哈希快速计算,这些位基本上对应于长度为该位的 k-mer。
我们在模拟和合成宏基因组实验的 NGS 数据上进行了多次实验,以评估相对于几种间隔种子计算每个读取中每个位置的哈希计算所需的时间。在我们的实验中,FISH 可以相对于传统方法提供 1.9x 到 6.03x 的加速,具体取决于间隔种子的结构。