Nasab Rezvan Rezaee, Mansourian Mahboubeh, Hassanzadeh Farshid, Shahlaei Mohsen
Department of Medicinal Chemistry, School of Pharmacy and Pharmaceutical Sciences, Lorestan University of Medical Sciences, Khorramabad, I.R. Iran.
Department of Medicinal Chemistry, School of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, I.R. Iran.
Res Pharm Sci. 2018 Dec;13(6):509-522. doi: 10.4103/1735-5362.245963.
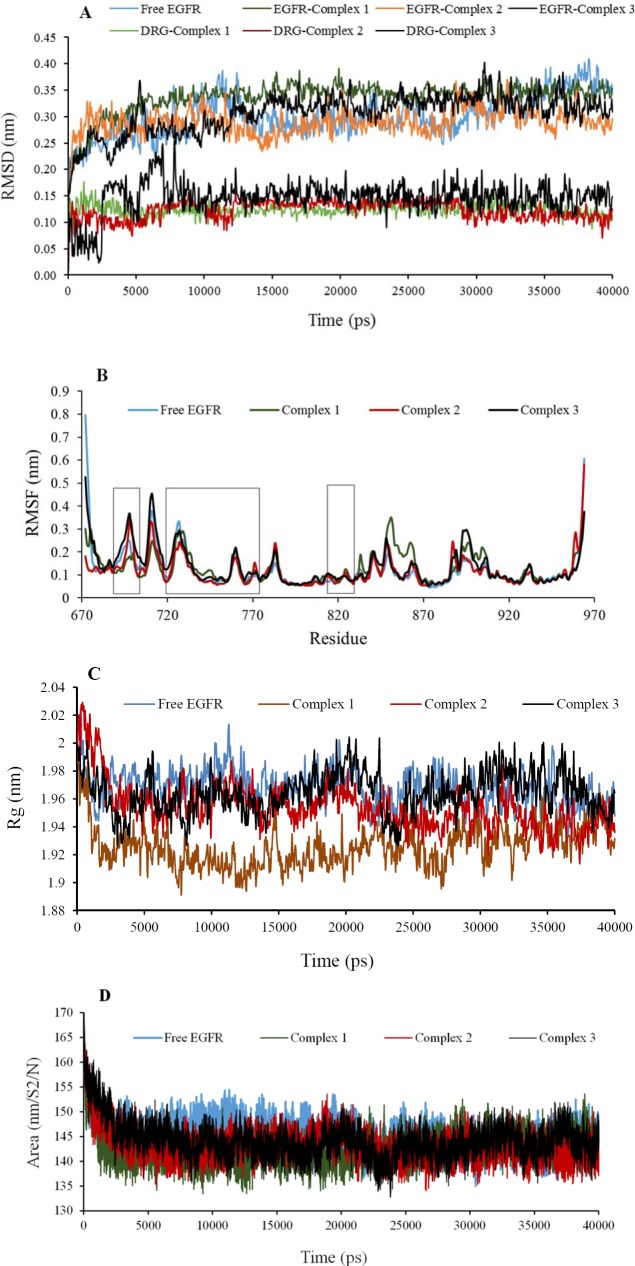
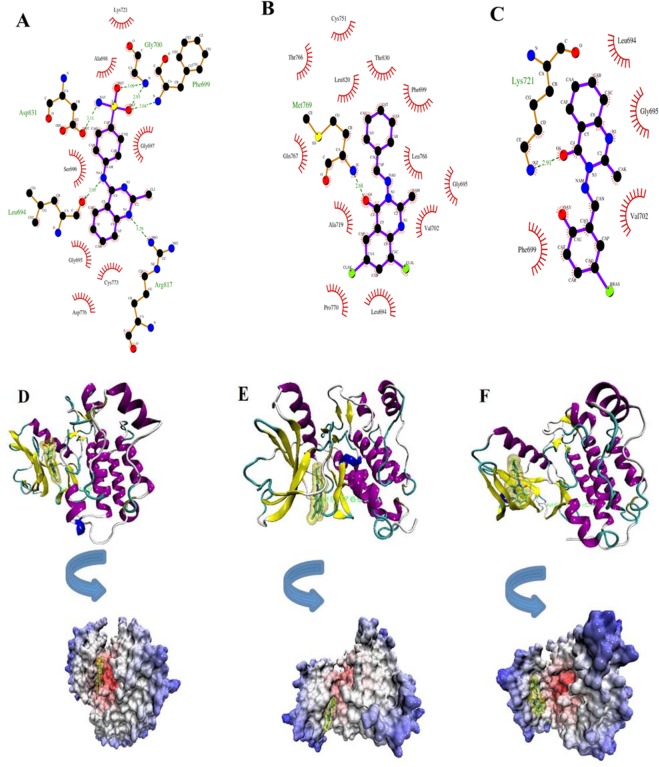
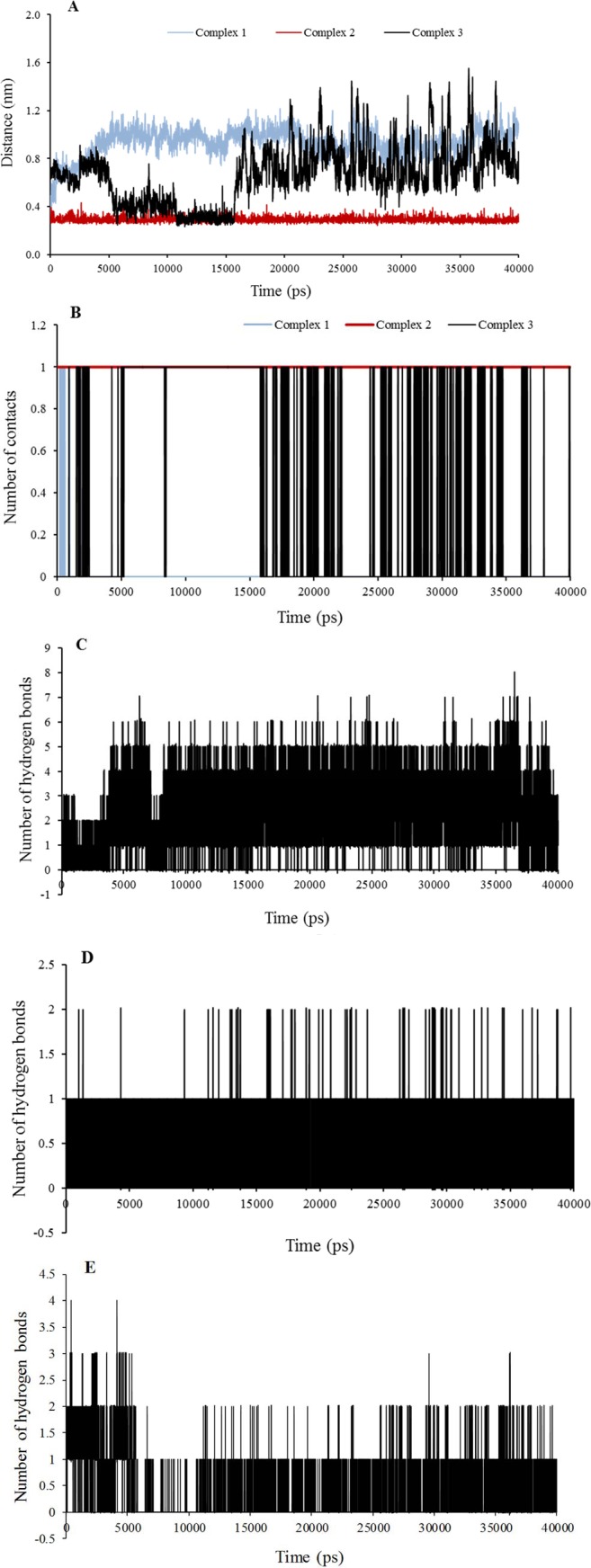
Quinazoline derivatives are potent inhibitors of human epidermal growth factor receptor (EGFR) as anticancer agents. In this study, the cytotoxic effects of a new series of synthesized quinazoline derivatives were evaluated using MTT assay against MCF-7 and HT-29 cell lines. Using molecular docking, the binding modes of all compounds were analyzed at the binding site of EGFR. Based on the results, the compounds L1, L2, L4, L5, L6, L7, L10, L15, and L18 may be promising EGFR inhibitors based on docking score and hydrogen bonds. Consistent with the experimental data, Met769 is recognized as a key residue in the binding of potential inhibitors. According to the MTT cytotoxicity assays, Lipinski's rule of five (RO5), absorption, distribution, metabolism, excretion, and toxicity (ADMET) parameters, and docking studies, three compounds L4, L15, and L10 with IC values of 80, 60, and 1 μM against the MCF-7 were selected for further comparative assessments. The dynamics of free EGFR, and selected ligand-EGFR complexes were investigated using molecular dynamics (MD) simulation studies. The results indicated that the three compounds bound to EGFR active site in a stable manner during the simulation through the formation of new hydrogen bonds with Phe699, Leu694, Gly700, Lys721, Met769, Arg817, and Asp831 with the superiority of compound L15. These features can promote future drug candidate designing to produce better derivatives in the search for the anticancer agents.
喹唑啉衍生物作为抗癌药物,是人类表皮生长因子受体(EGFR)的有效抑制剂。在本研究中,使用MTT法对一系列新合成的喹唑啉衍生物对MCF-7和HT-29细胞系的细胞毒性作用进行了评估。通过分子对接,分析了所有化合物在EGFR结合位点的结合模式。根据结果,基于对接分数和氢键,化合物L1、L2、L4、L5、L6、L7、L10、L15和L18可能是有前景的EGFR抑制剂。与实验数据一致,Met769被认为是潜在抑制剂结合中的关键残基。根据MTT细胞毒性试验、Lipinski的五规则(RO5)、吸收、分布、代谢、排泄和毒性(ADMET)参数以及对接研究,选择了对MCF-7的IC值分别为80、60和1 μM的三种化合物L4、L15和L10进行进一步的比较评估。使用分子动力学(MD)模拟研究了游离EGFR以及选定的配体-EGFR复合物的动力学。结果表明,在模拟过程中,这三种化合物通过与Phe699、Leu694、Gly700、Lys721、Met769、Arg817和Asp831形成新的氢键,以稳定的方式结合到EGFR活性位点,其中化合物L15表现更优。这些特性有助于未来设计更好的候选药物,以开发出更优的抗癌药物衍生物。