Division of Gastroenterology, Hepatology, and Nutrition, Department of Pediatrics, University of California, San Diego School of Medicine, La Jolla, California; Department of Gastroenterology, Rady Children's Hospital San Diego, San Diego, California.
The Jackson Laboratory for Genomic Medicine, Farmington, Connecticut.
Gastroenterology. 2019 Oct;157(4):1109-1122. doi: 10.1053/j.gastro.2019.06.028. Epub 2019 Jun 27.
BACKGROUND & AIMS: The intestinal microbiome might affect the development and severity of nonalcoholic fatty liver disease (NAFLD). We analyzed microbiomes of children with and without NAFLD.
We performed a prospective, observational, cross-sectional study of 87 children (age range, 8-17 years) with biopsy-proven NAFLD and 37 children with obesity without NAFLD (controls). Fecal samples were collected and microbiome composition and functions were assessed using 16S ribosomal RNA amplicon sequencing and metagenomic shotgun sequencing. Microbial taxa were identified using zero-inflated negative binomial modeling. Genes contributing to bacterial pathways were identified using gene set enrichment analysis.
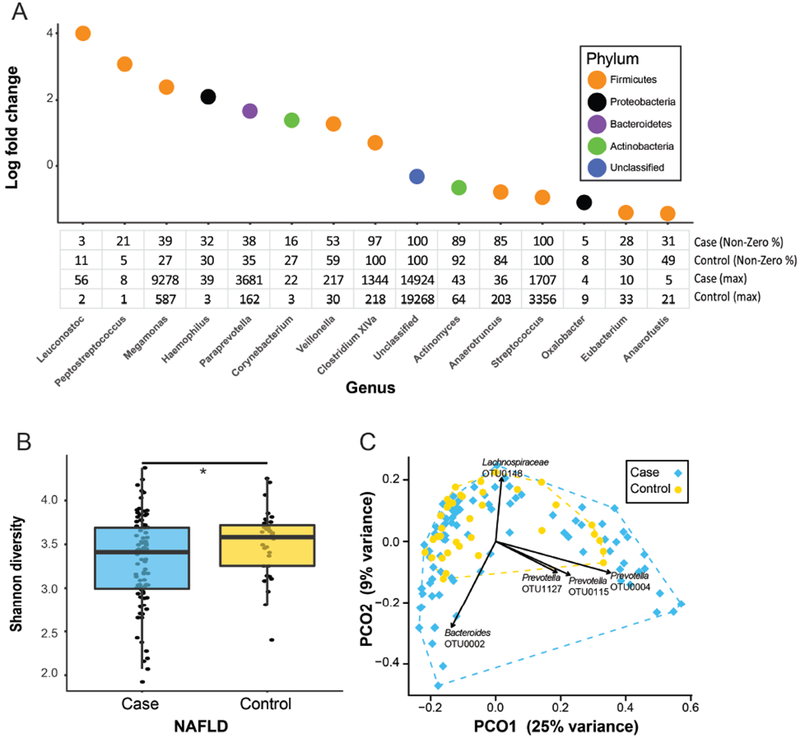
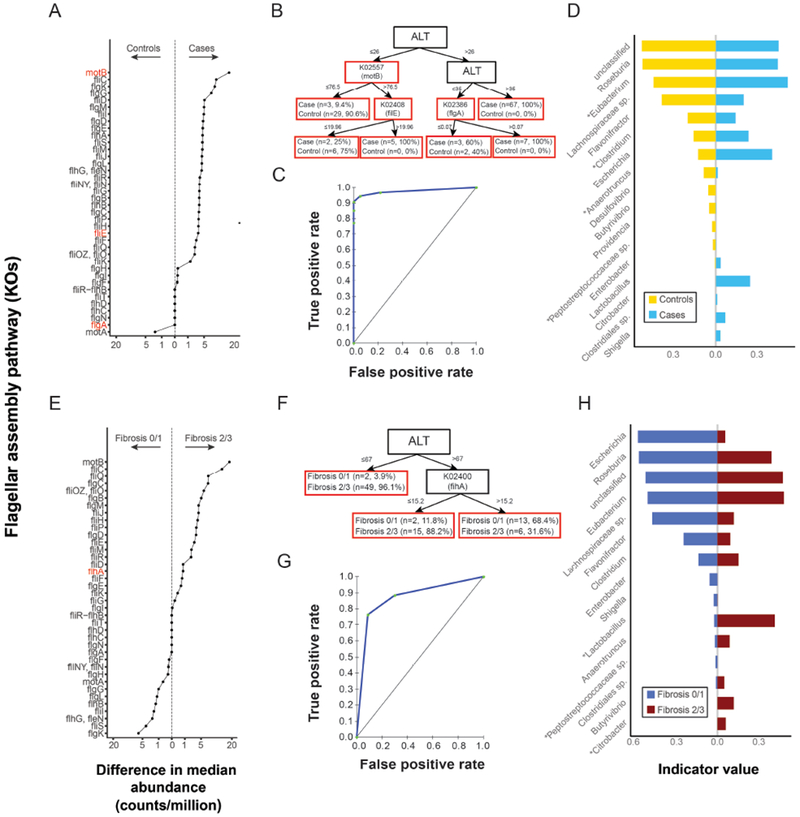
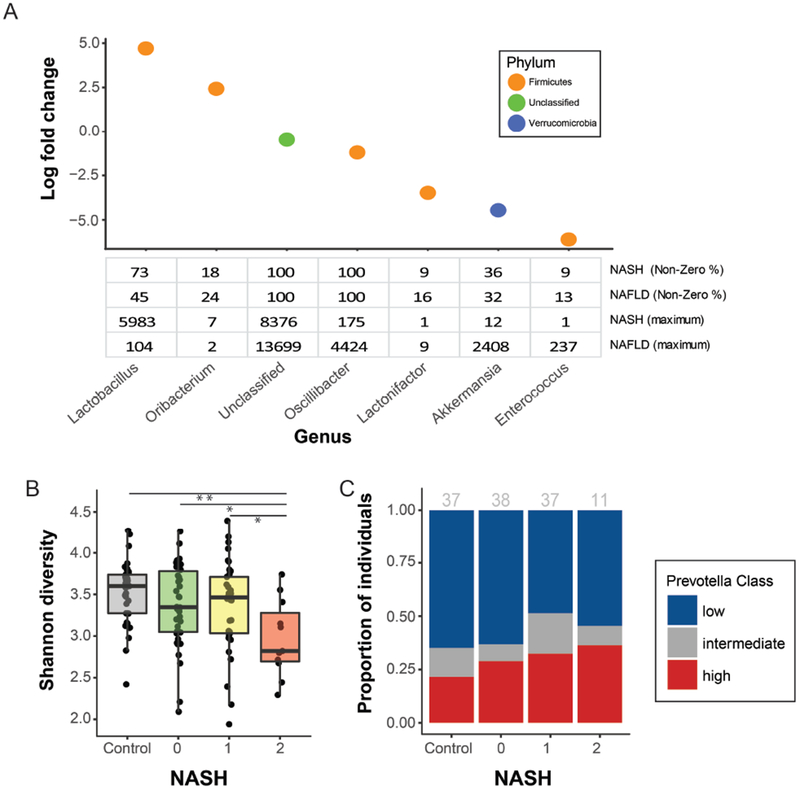
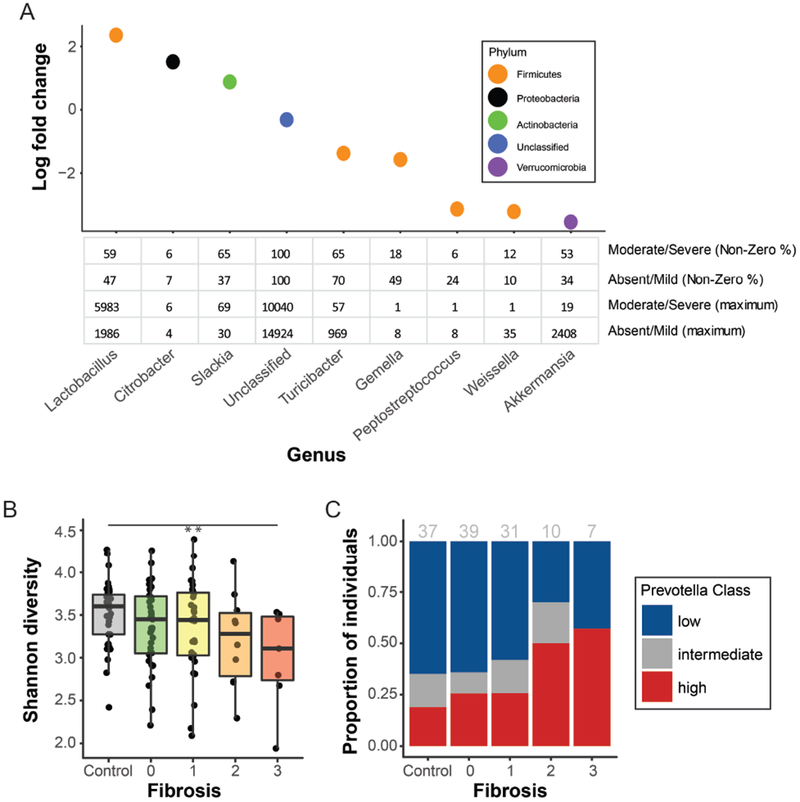
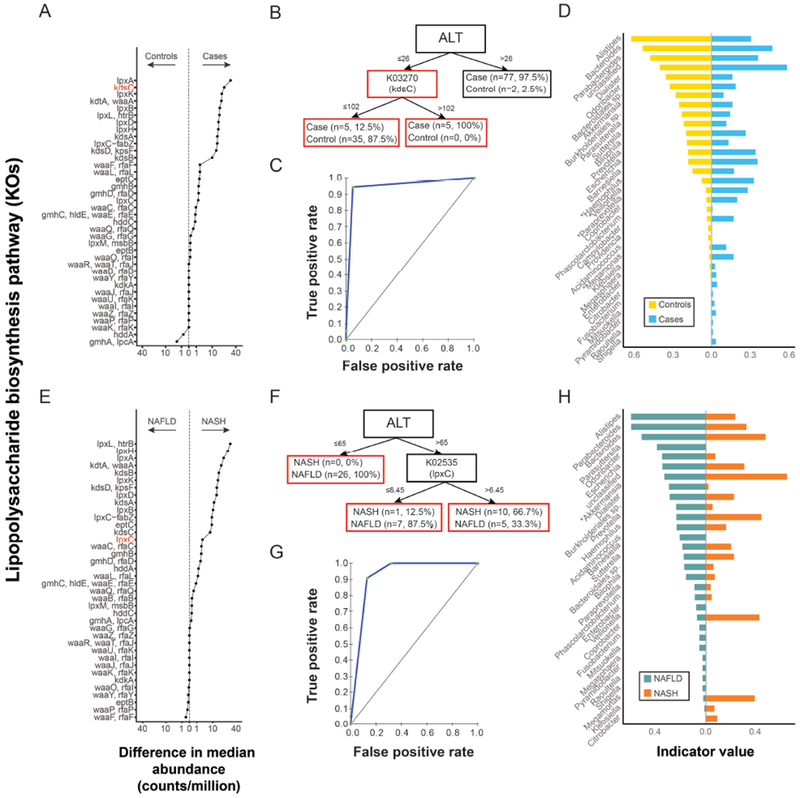
Fecal microbiomes of children with NAFLD had lower α-diversity than those of control children (3.32 vs 3.52, P = .016). Fecal microbiomes from children with nonalcoholic steatohepatitis (NASH) had the lowest α-diversity (control, 3.52; NAFLD, 3.36; borderline NASH, 3.37; NASH, 2.97; P = .001). High abundance of Prevotella copri was associated with more severe fibrosis (P = .036). Genes for lipopolysaccharide biosynthesis were enriched in microbiomes from children with NASH (P < .001). Classification and regression tree model with level of alanine aminotransferase and relative abundance of the lipopolysaccharide pathway gene encoding 3-deoxy-d-manno-octulosonate 8-phosphate-phosphatase identified patients with NASH with an area under the receiver operating characteristic curve value of 0.92. Genes involved in flagellar assembly were enriched in the fecal microbiomes of patients with moderate to severe fibrosis (P < .001). Classification and regression tree models based on level of alanine aminotransferase and abundance of genes encoding flagellar biosynthesis protein had good accuracy for identifying case children with moderate to severe fibrosis (area under the receiver operating characteristic curve, 0.87).
In an analysis of fecal microbiomes of children with NAFLD, we associated NAFLD and NASH with intestinal dysbiosis. NAFLD and its severity were associated with greater abundance of genes encoding inflammatory bacterial products. Alterations to the intestinal microbiome might contribute to the pathogenesis of NAFLD and be used as markers of disease or severity.
肠道微生物群可能会影响非酒精性脂肪性肝病(NAFLD)的发生和严重程度。我们分析了患有和不患有 NAFLD 的儿童的微生物组。
我们对 87 名经活检证实患有 NAFLD 的儿童(年龄 8-17 岁)和 37 名患有非酒精性脂肪肝的肥胖儿童(对照组)进行了前瞻性、观察性、横断面研究。收集粪便样本,使用 16S 核糖体 RNA 扩增子测序和宏基因组鸟枪法测序评估微生物组组成和功能。使用零膨胀负二项式模型鉴定微生物分类群。使用基因集富集分析鉴定与细菌途径相关的基因。
患有 NAFLD 的儿童的粪便微生物组的 α-多样性低于对照组儿童(3.32 对 3.52,P=0.016)。非酒精性脂肪性肝炎(NASH)患儿的粪便微生物组的 α-多样性最低(对照组 3.52;NAFLD 3.36;边缘性 NASH 3.37;NASH 2.97;P=0.001)。Prevotella copri 的丰度与更严重的纤维化相关(P=0.036)。NASH 患儿的微生物组中富含脂多糖生物合成基因(P<0.001)。基于丙氨酸氨基转移酶水平和内毒素途径基因相对丰度的分类和回归树模型,可识别出 NASH 患者,其受试者工作特征曲线下面积值为 0.92。在中重度纤维化患者的粪便微生物组中,富集了参与鞭毛组装的基因(P<0.001)。基于丙氨酸氨基转移酶水平和编码鞭毛生物合成蛋白的基因丰度的分类和回归树模型,对中重度纤维化患者的识别具有良好的准确性(受试者工作特征曲线下面积,0.87)。
在对患有 NAFLD 的儿童的粪便微生物组进行分析时,我们将 NAFLD 和 NASH 与肠道菌群失调相关联。NAFLD 及其严重程度与编码炎症性细菌产物的基因丰度增加有关。肠道微生物组的改变可能有助于 NAFLD 的发病机制,并可作为疾病或严重程度的标志物。