Pathogen Genomics Center, National Institute of Infectious Diseases, Tokyo, Japan.
Department of Infectious Diseases, Tokyo Women's Medical University, Tokyo, Japan.
PLoS One. 2019 Oct 9;14(10):e0223433. doi: 10.1371/journal.pone.0223433. eCollection 2019.
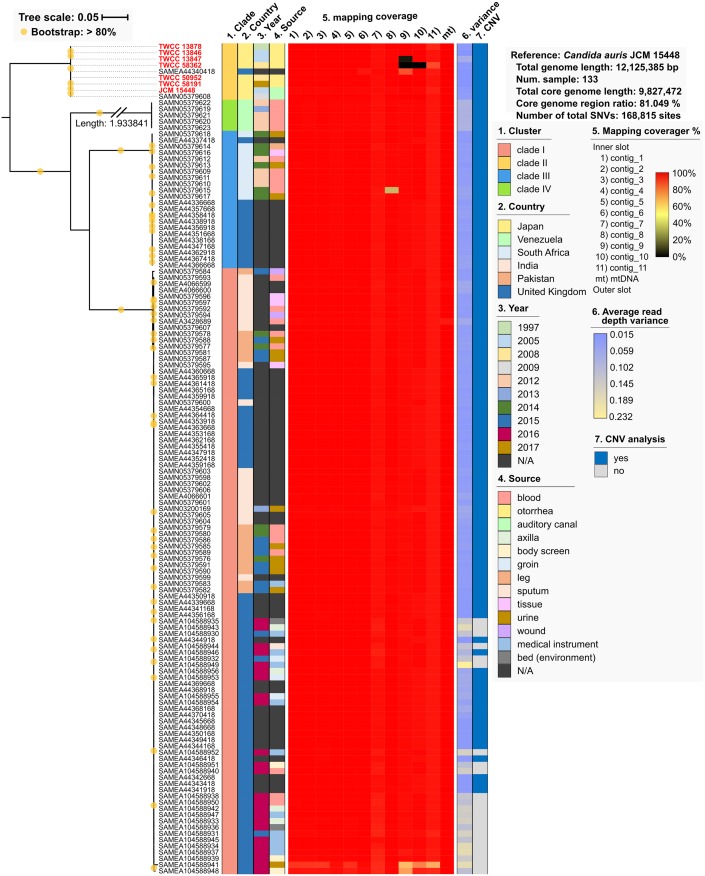
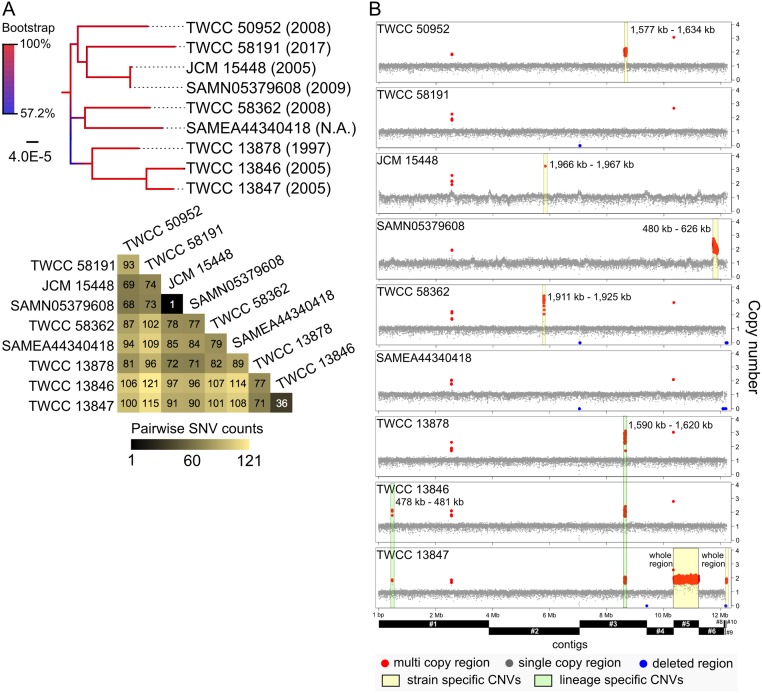
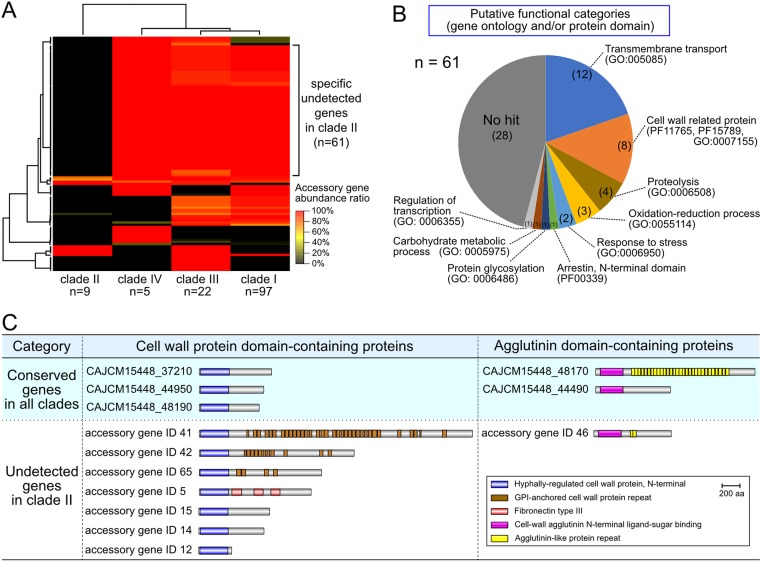
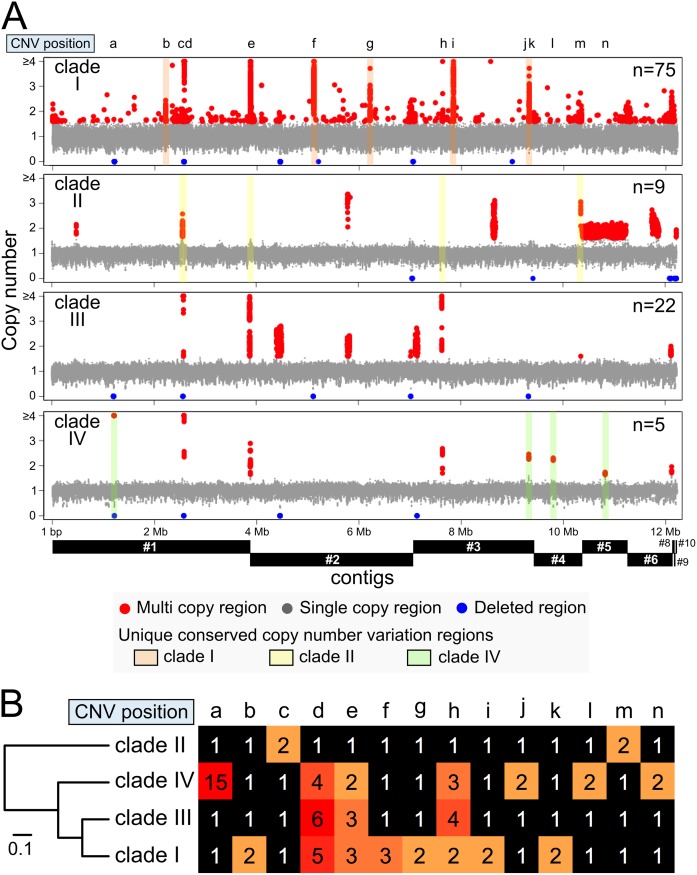
Candida auris is an invasive and multidrug-resistant ascomycetous yeast that is under global surveillance. All clinical cases of C. auris infection diagnosed from 1997 to 2019 in Japan were non-invasive and sporadic otitis media cases. In the present study, we performed whole-genome sequencing of seven C. auris strains isolated from patients with otitis media in Japan, all of which belonged to clade II. Comparative genome analysis using the high-quality draft genome sequences JCM 15448T revealed that single nucleotide variations (SNVs), clade-specific accessory genes, and copy number variations (CNVs) were identified in each C. auris clade. A total of 61 genes involved in cell wall and stress response-related functions was absent in clade II, and the pattern of conserved CNVs in each clade was more stable in clade II than in other clades. Our data suggest that the genomic structural diversity is stable in C. auris isolated from each biogeographic location, and Japanese strains isolated from patients with otitis media might belong to an ancestral type of C. auris. One Japanese strain, TWCC 58362, with reduced susceptibility to fluconazole, exhibited no mutation in ergosterol biosynthesis-related genes (ERG). However, TWCC 58362-specific variations, including SNVs, indels, and CNVs were detected, suggesting that gene duplication events in C. auris might contribute to antifungal drug resistance. Taken together, we demonstrated that genomic structural variations in C. auris could correlate to geographical dissemination, epidemiology, lesions in the host, and antifungal resistance.
耳念珠菌是一种具有侵袭性和多药耐药性的子囊菌,目前正在全球范围内受到监测。1997 年至 2019 年在日本诊断的所有耳念珠菌感染临床病例均为非侵袭性和散发性中耳炎病例。在本研究中,我们对从日本中耳炎患者中分离出的 7 株耳念珠菌菌株进行了全基因组测序,这些菌株均属于 II 型分支。使用高质量的 JCM 15448T 草图基因组序列进行比较基因组分析表明,每个耳念珠菌分支都存在单核苷酸变异(SNVs)、分支特异性辅助基因和拷贝数变异(CNVs)。细胞壁和应激反应相关功能的 61 个基因在 II 型分支中缺失,每个分支中保守 CNVs 的模式在 II 型分支中比其他分支更稳定。我们的数据表明,从每个生物地理区域分离出的耳念珠菌的基因组结构多样性是稳定的,从中耳炎患者中分离出的日本菌株可能属于耳念珠菌的原始类型。一株对氟康唑敏感性降低的日本菌株 TWCC 58362 未在与麦角固醇生物合成相关的基因(ERG)中检测到突变。然而,在 TWCC 58362 中检测到特定的变异,包括 SNVs、插入缺失和 CNVs,表明耳念珠菌中的基因重复事件可能导致抗真菌药物耐药性。总之,我们证明了耳念珠菌的基因组结构变异可能与地理传播、流行病学、宿主病变和抗真菌耐药性相关。