Hereditary Cancer Group, Program for Predictive and Personalized Medicine of Cancer, Germans Trias i Pujol Research Institute (PMPPC-IGTP), Campus Can Ruti, Badalona, Spain.
Hereditary Cancer Program, Joint Program on Hereditary Cancer, Catalan Institute of Oncology, Institut d'Investigació Biomèdica de Bellvitge-IDIBELL, L'Hospitalet de Llobregat, Barcelona, Spain.
Eur J Hum Genet. 2020 Dec;28(12):1645-1655. doi: 10.1038/s41431-020-0675-z. Epub 2020 Jun 19.
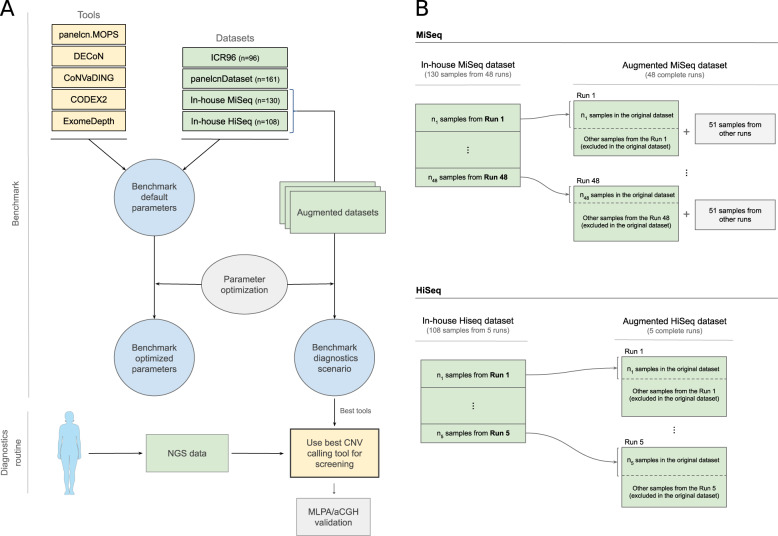
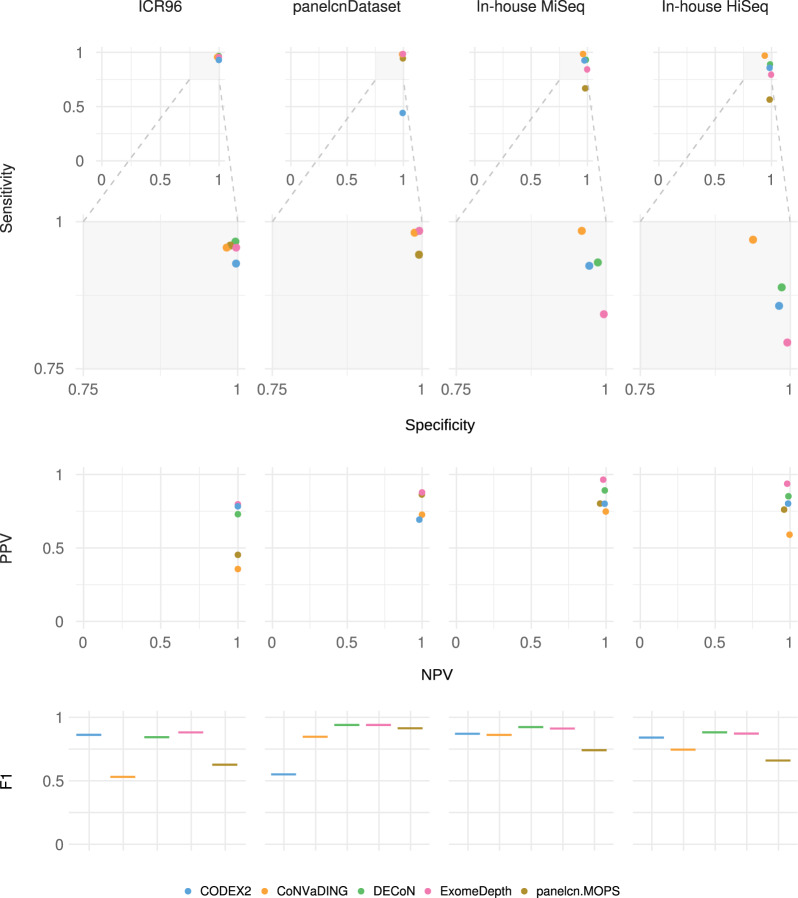
Although germline copy-number variants (CNVs) are the genetic cause of multiple hereditary diseases, detecting them from targeted next-generation sequencing data (NGS) remains a challenge. Existing tools perform well for large CNVs but struggle with single and multi-exon alterations. The aim of this work is to evaluate CNV calling tools working on gene panel NGS data and their suitability as a screening step before orthogonal confirmation in genetic diagnostics strategies. Five tools (DECoN, CoNVaDING, panelcn.MOPS, ExomeDepth, and CODEX2) were tested against four genetic diagnostics datasets (two in-house and two external) for a total of 495 samples with 231 single and multi-exon validated CNVs. The evaluation was performed using the default and sensitivity-optimized parameters. Results showed that most tools were highly sensitive and specific, but the performance was dataset dependant. When evaluating them in our diagnostics scenario, DECoN and panelcn.MOPS detected all CNVs with the exception of one mosaic CNV missed by DECoN. However, DECoN outperformed panelcn.MOPS specificity achieving values greater than 0.90 when using the optimized parameters. In our in-house datasets, DECoN and panelcn.MOPS showed the highest performance for CNV screening before orthogonal confirmation. Benchmarking and optimization code is freely available at https://github.com/TranslationalBioinformaticsIGTP/CNVbenchmarkeR .
尽管种系拷贝数变异(CNVs)是多种遗传性疾病的遗传原因,但从靶向下一代测序数据(NGS)中检测它们仍然是一个挑战。现有的工具在处理大的 CNVs 时表现良好,但在处理单和多外显子改变时则存在困难。本工作的目的是评估在基因面板 NGS 数据上工作的 CNV 调用工具及其作为遗传诊断策略中正交确认之前的筛选步骤的适用性。使用默认和灵敏度优化参数对五种工具(DECon、CoNVaDING、panelcn.MOPS、ExomeDepth 和 CODEX2)进行了测试,共涉及 495 个样本,其中 231 个为经单和多外显子验证的 CNVs。结果表明,大多数工具具有高度的敏感性和特异性,但性能取决于数据集。在我们的诊断方案中评估它们时,DECon 和 panelcn.MOPS 检测到了所有的 CNV,除了一个 DECoN 漏掉的嵌合体 CNV。然而,当使用优化参数时,DECon 的特异性优于 panelcn.MOPS,达到了大于 0.90 的值。在我们的内部数据集,DECon 和 panelcn.MOPS 显示出在正交确认之前进行 CNV 筛选的最高性能。基准测试和优化代码可在 https://github.com/TranslationalBioinformaticsIGTP/CNVbenchmarkeR 上免费获得。