Department of Genetics and Genomic Sciences and Icahn Institute for Data Science and Genomic Technology, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Department of Environmental Medicine and Public Health, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Metabolomics. 2020 Oct 21;16(11):117. doi: 10.1007/s11306-020-01738-3.
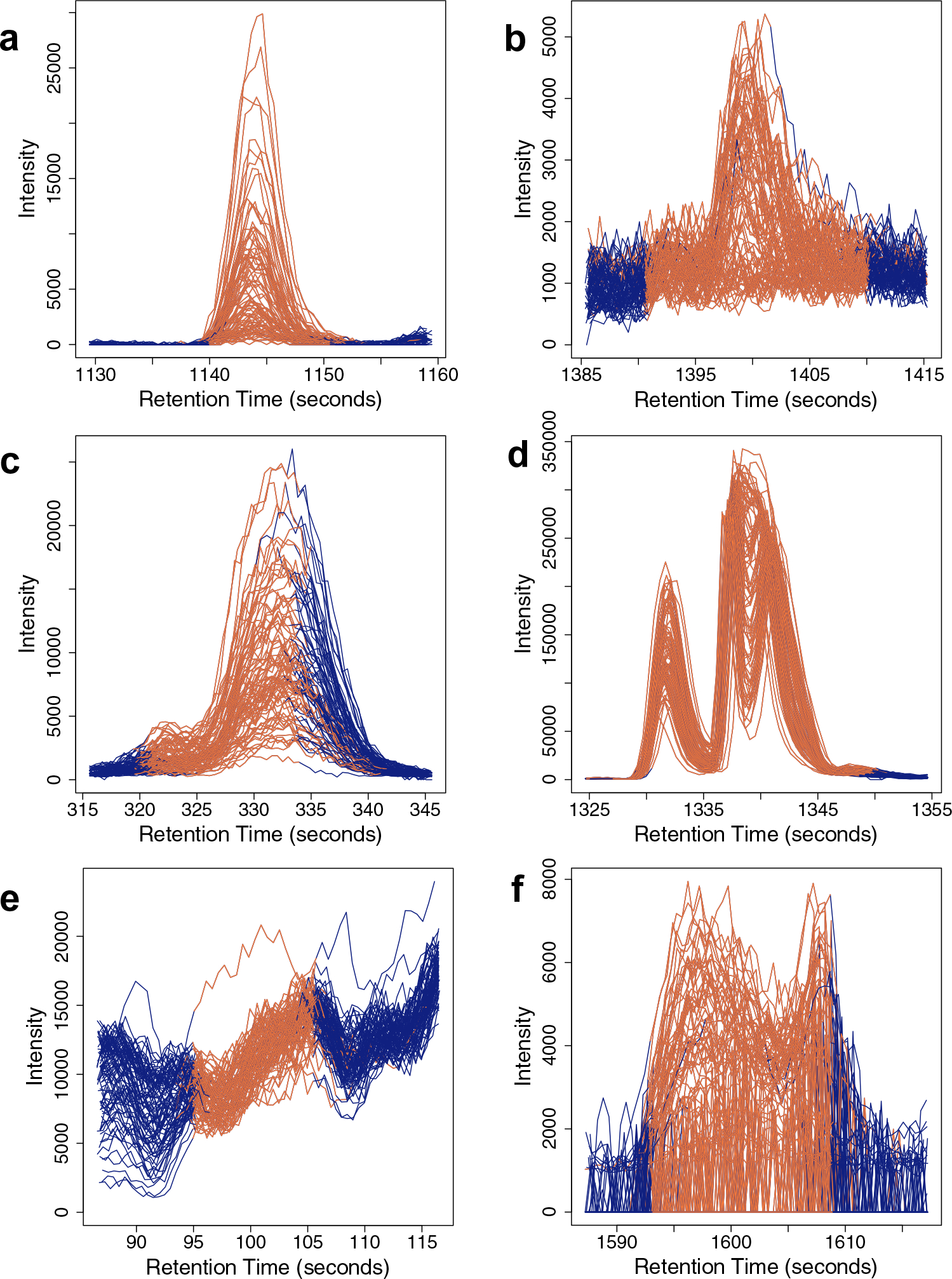
Despite the availability of several pre-processing software, poor peak integration remains a prevalent problem in untargeted metabolomics data generated using liquid chromatography high-resolution mass spectrometry (LC-MS). As a result, the output of these pre-processing software may retain incorrectly calculated metabolite abundances that can perpetuate in downstream analyses.
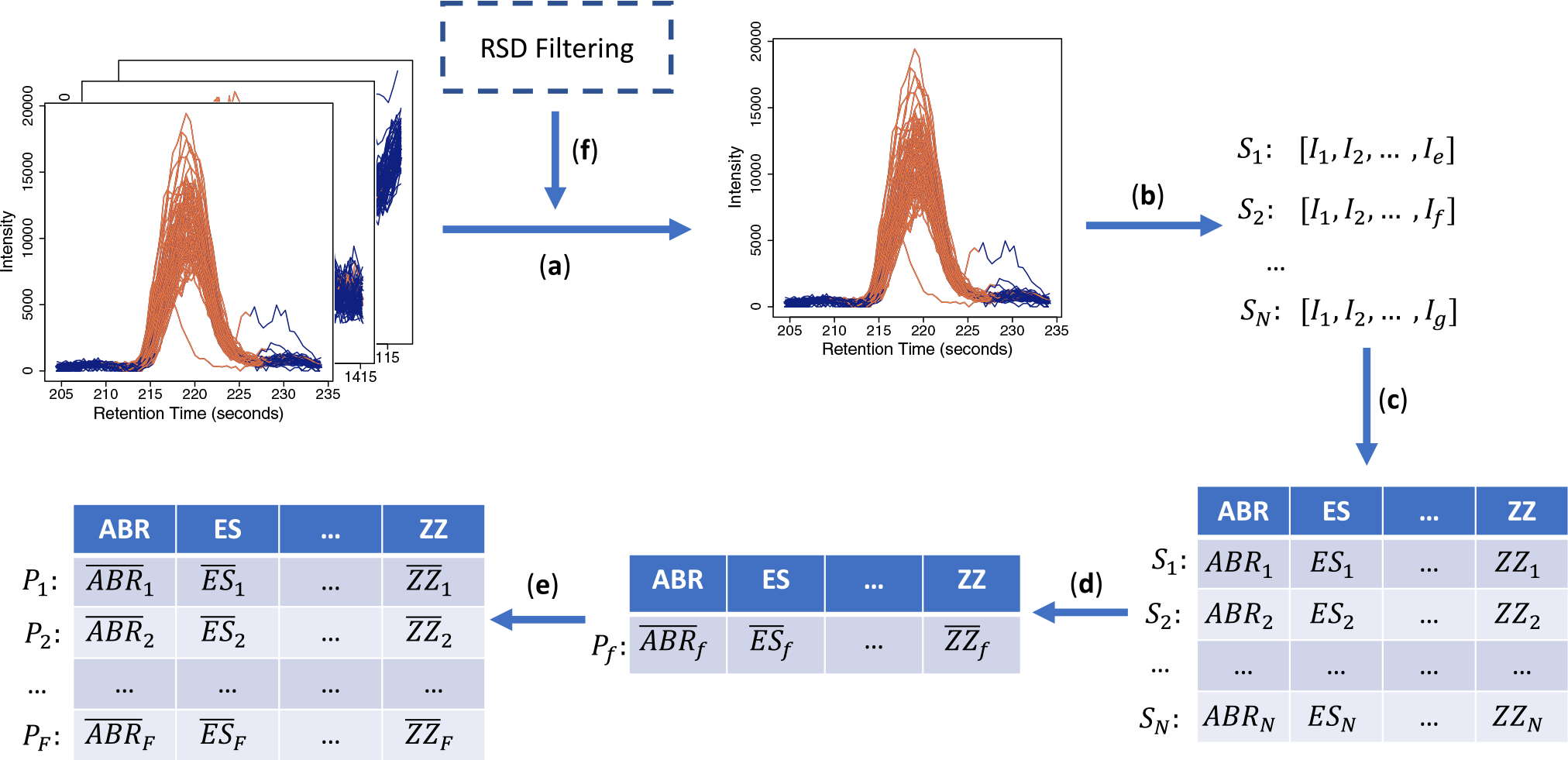
To address this problem, we propose a computational methodology that combines machine learning and peak quality metrics to filter out low quality peaks.
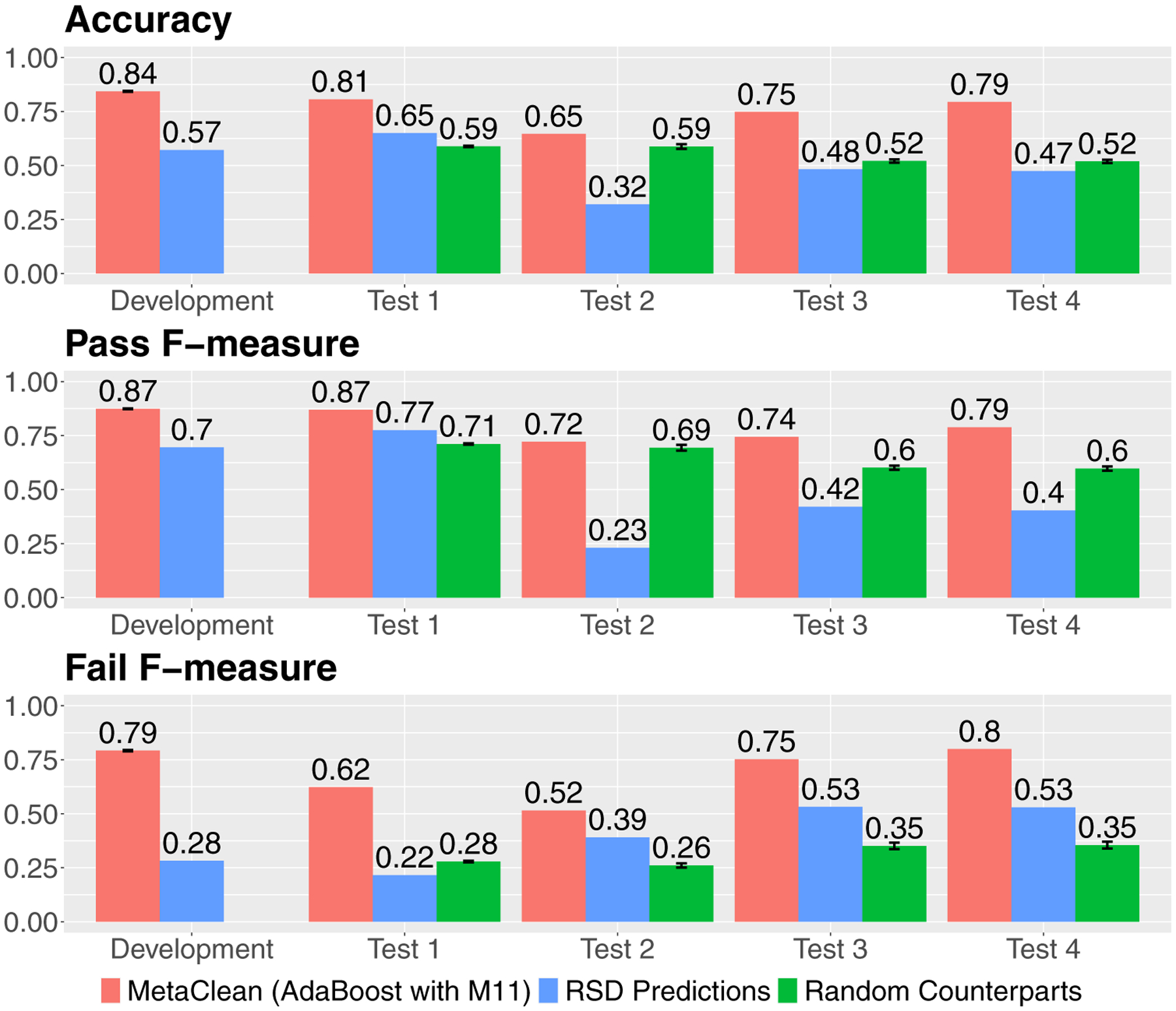
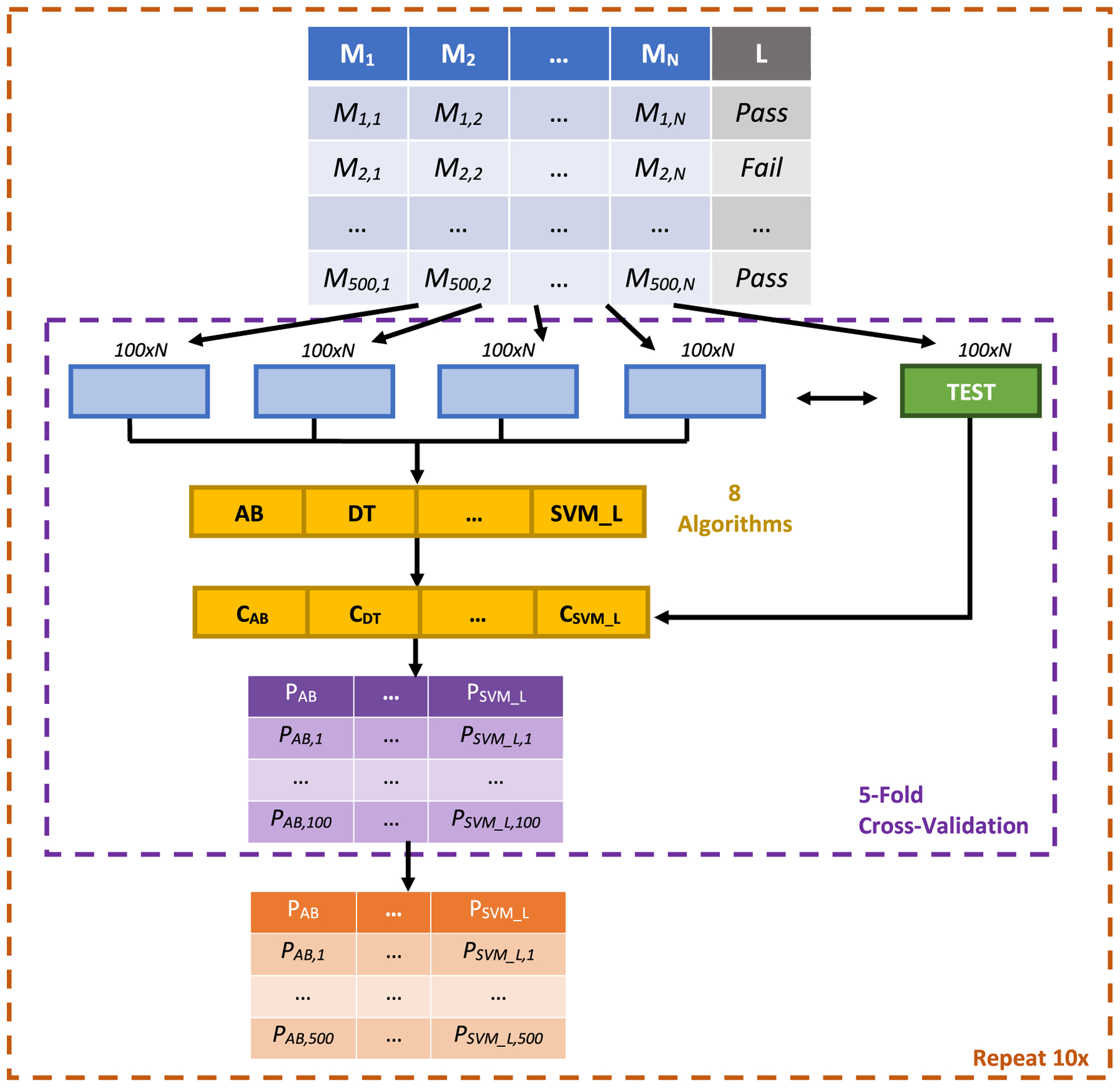
Specifically, we comprehensively and systematically compared the performance of 24 different classifiers generated by combining eight classification algorithms and three sets of peak quality metrics on the task of distinguishing reliably integrated peaks from poorly integrated ones. These classifiers were compared to using a residual standard deviation (RSD) cut-off in pooled quality-control (QC) samples, which aims to remove peaks with analytical error.
The best performing classifier was found to be a combination of the AdaBoost algorithm and a set of 11 peak quality metrics previously explored in untargeted metabolomics and proteomics studies. As a complementary approach, applying our framework to peaks retained after filtering by 30% RSD across pooled QC samples was able to further distinguish poorly integrated peaks that were not removed from filtering alone. An R implementation of these classifiers and the overall computational approach is available as the MetaClean package at https://CRAN.R-project.org/package=MetaClean .
Our work represents an important step forward in developing an automated tool for filtering out unreliable peak integrations in untargeted LC-MS metabolomics data.
尽管有多种预处理软件可供使用,但在使用液相色谱高分辨率质谱(LC-MS)生成的非靶向代谢组学数据中,峰积分仍然是一个普遍存在的问题。因此,这些预处理软件的输出可能会保留不正确计算的代谢物丰度,这些丰度可能会在下游分析中持续存在。
为了解决这个问题,我们提出了一种结合机器学习和峰质量指标的计算方法,以过滤出低质量的峰。
具体来说,我们全面系统地比较了 24 种不同分类器的性能,这些分类器是通过将八种分类算法和三套峰质量指标结合起来,用于区分可靠积分峰和积分不良峰。这些分类器与使用综合质量控制(QC)样本中的剩余标准差(RSD)截止值进行比较,其目的是去除具有分析误差的峰。
发现性能最佳的分类器是一种组合,结合了 AdaBoost 算法和一套 11 个峰质量指标,这些指标之前在非靶向代谢组学和蛋白质组学研究中进行了探索。作为一种补充方法,将我们的框架应用于通过 30%RSD 过滤后保留的峰,能够进一步区分那些仅通过过滤无法去除的积分不良峰。这些分类器和整体计算方法的 R 实现可作为 MetaClean 软件包在 https://CRAN.R-project.org/package=MetaClean 上获得。
我们的工作在开发用于过滤非靶向 LC-MS 代谢组学数据中不可靠峰积分的自动化工具方面迈出了重要的一步。