Chien Li-Chu
Center for Fundamental Science, Kaohsiung Medical University, Kaohsiung, Taiwan.
Comput Math Methods Med. 2021 Feb 9;2021:8812282. doi: 10.1155/2021/8812282. eCollection 2021.
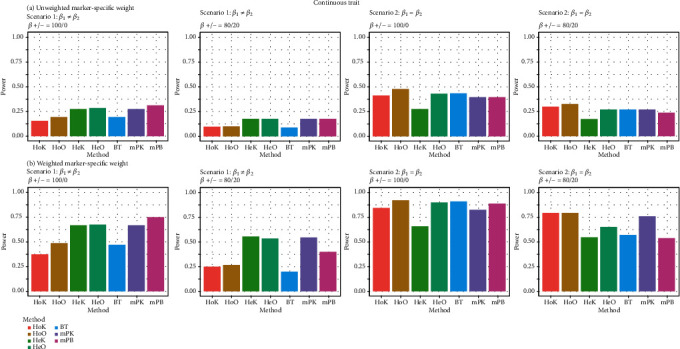
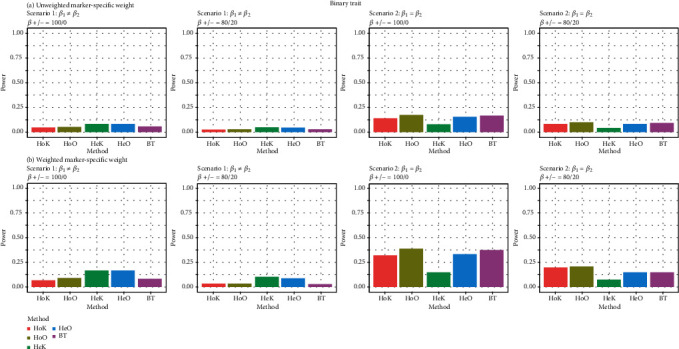
In genetic association analysis, several relevant phenotypes or multivariate traits with different types of components are usually collected to study complex or multifactorial diseases. Over the past few years, jointly testing for association between multivariate traits and multiple genetic variants has become more popular because it can increase statistical power to identify causal genes in pedigree- or population-based studies. However, most of the existing methods mainly focus on testing genetic variants associated with multiple continuous phenotypes. In this investigation, we develop a framework for identifying the pleiotropic effects of genetic variants on multivariate traits by using collapsing and kernel methods with pedigree- or population-structured data. The proposed framework is applicable to the burden test, the kernel test, and the omnibus test for autosomes and the X chromosome. The proposed multivariate trait association methods can accommodate continuous phenotypes or binary phenotypes and further can adjust for covariates. Simulation studies show that the performance of our methods is satisfactory with respect to the empirical type I error rates and power rates in comparison with the existing methods.
在基因关联分析中,通常会收集几种相关的表型或具有不同类型成分的多变量性状,以研究复杂或多因素疾病。在过去几年中,对多变量性状和多个基因变异之间的关联进行联合检验变得越来越普遍,因为它可以提高在基于家系或人群的研究中识别因果基因的统计效力。然而,现有的大多数方法主要集中于检验与多个连续表型相关的基因变异。在本研究中,我们开发了一个框架,通过使用基于家系或人群结构数据的合并和核方法来识别基因变异对多变量性状的多效性作用。所提出的框架适用于常染色体和X染色体的负担检验、核检验和综合检验。所提出的多变量性状关联方法可以处理连续表型或二元表型,并且还可以对协变量进行调整。模拟研究表明,与现有方法相比,我们的方法在经验I型错误率和检验功效方面的表现令人满意。