Department of Computer Science, University of Illinois Urbana-Champaign, Urbana, IL 61820, USA.
Bioinformatics. 2023 Jan 1;39(1). doi: 10.1093/bioinformatics/btad007.
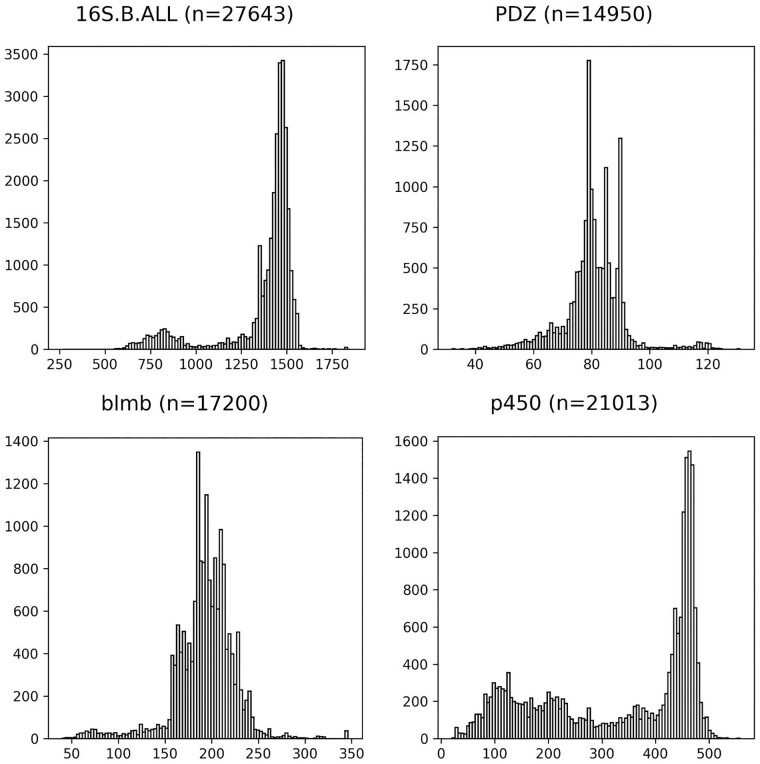
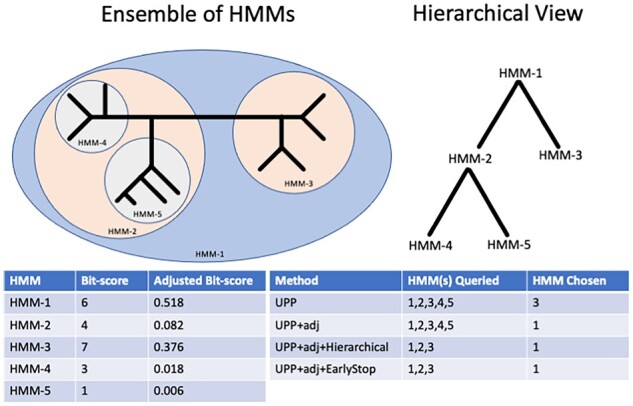
Multiple sequence alignment (MSA) is a basic step in many bioinformatics pipelines. However, achieving highly accurate alignments on large datasets, especially those with sequence length heterogeneity, is a challenging task. Ultra-large multiple sequence alignment using Phylogeny-aware Profiles (UPP) is a method for MSA estimation that builds an ensemble of Hidden Markov Models (eHMM) to represent an estimated alignment on the full-length sequences in the input, and then adds the remaining sequences into the alignment using selected HMMs in the ensemble. Although UPP provides good accuracy, it is computationally intensive on large datasets.
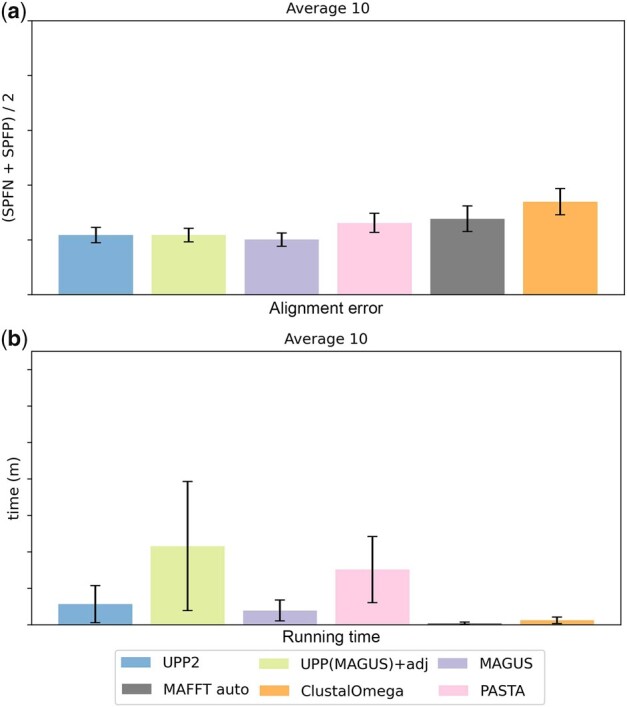
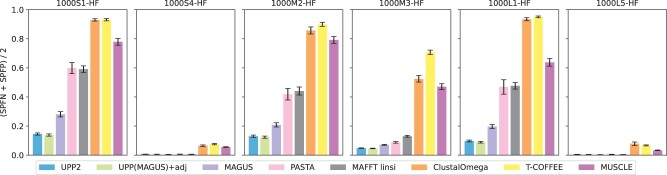
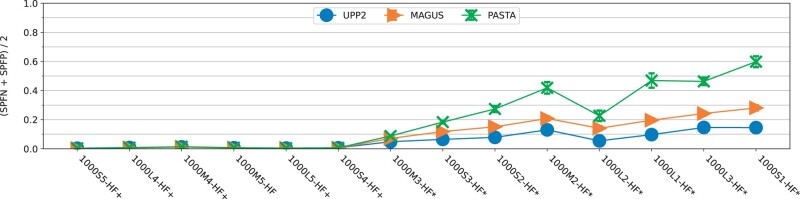
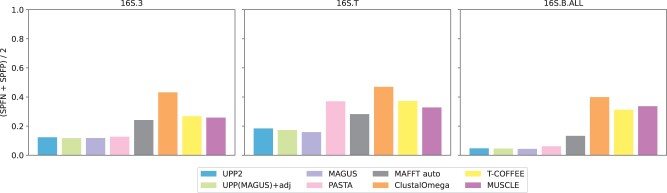
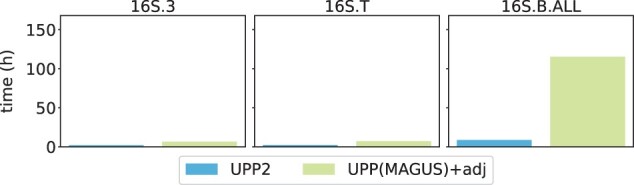
We present UPP2, a direct improvement on UPP. The main advance is a fast technique for selecting HMMs in the ensemble that allows us to achieve the same accuracy as UPP but with greatly reduced runtime. We show that UPP2 produces more accurate alignments compared to leading MSA methods on datasets exhibiting substantial sequence length heterogeneity and is among the most accurate otherwise.
https://github.com/gillichu/sepp.
Supplementary data are available at Bioinformatics online.
多序列比对(MSA)是许多生物信息学流程的基本步骤。然而,在大型数据集上实现高度准确的比对,特别是那些具有序列长度异质性的数据集,是一项具有挑战性的任务。使用 Phylogeny-aware Profiles(UPP)进行超大型多序列比对是一种 MSA 估计方法,它构建了一个隐马尔可夫模型(HMM)的集合来表示输入的全长序列上的估计比对,然后使用集合中的选定 HMM 将其余序列添加到比对中。尽管 UPP 提供了很好的准确性,但在大型数据集上计算量很大。
我们提出了 UPP2,这是 UPP 的直接改进。主要的进展是一种在集合中选择 HMM 的快速技术,它允许我们实现与 UPP 相同的准确性,但运行时间大大缩短。我们表明,与具有大量序列长度异质性的数据集上的领先 MSA 方法相比,UPP2 产生了更准确的比对,并且在其他方面也是最准确的之一。
https://github.com/gillichu/sepp。
补充数据可在 Bioinformatics 在线获得。