Ekici Arif B, Oezbey Sevinc, Fuchs Christina, Nelis Eva, Van Broeckhoven Christine, Schachner Melitta, Rautenstrauss Bernd
Institute of Human Genetics, Friedrich-Alexander-University, Erlangen, Germany.
BMC Cell Biol. 2002 Nov 26;3:29. doi: 10.1186/1471-2121-3-29.
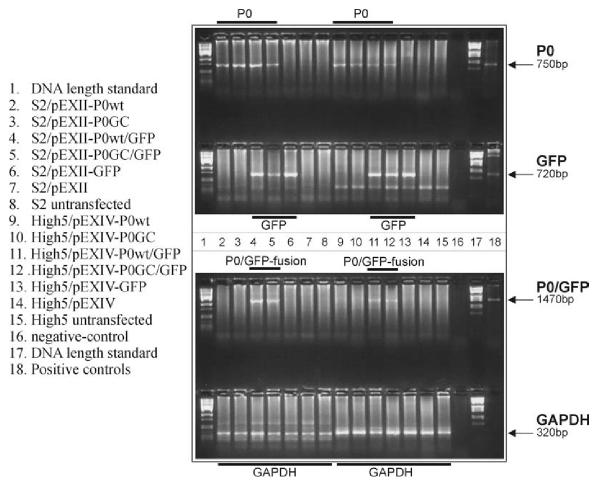
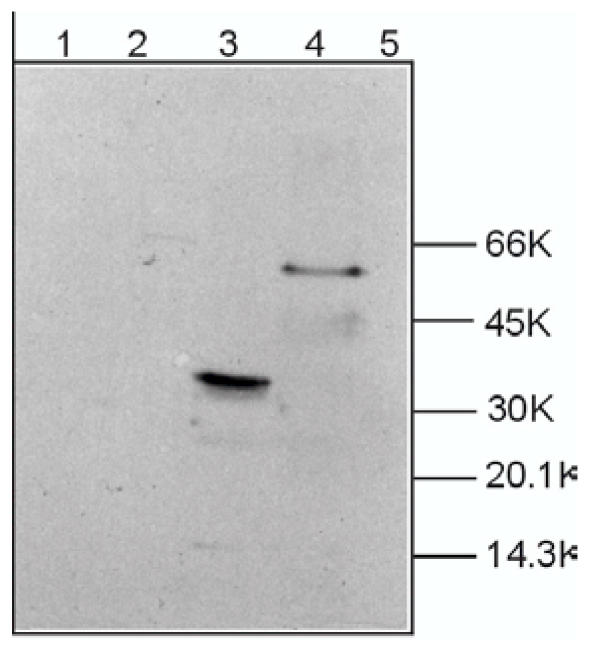
Mutations in P0, the major protein of the myelin sheath in peripheral nerves, cause the inherited peripheral neuropathies Charcot-Marie-Tooth disease type 1B (CMT1B), Dejerine-Sottas syndrome (DSS) and congenital hypomyelination (CH). We reported earlier a de novo insertional mutation c.662_663GC (Ala221fs) in a DSS patient. The c.662_663GC insertion results in a frame shift mutation Ala221fs altering the C-terminal amino acid sequence. The adhesion-relevant intracellular RSTK domain is replaced by a sequence similar to Na+/K+ ATPase. To further clarify the molecular disease mechanisms in this sporadic patient we constructed wild type P0 and the c.662_663GC mutant expression cassettes by site-specific mutagenesis and transfected the constructs into insect cells (S2, High5). To trace the effects in live cells, green fluorescent protein (GFP) has been added to the carboxyterminus of the wild type and mutated P0 protein.
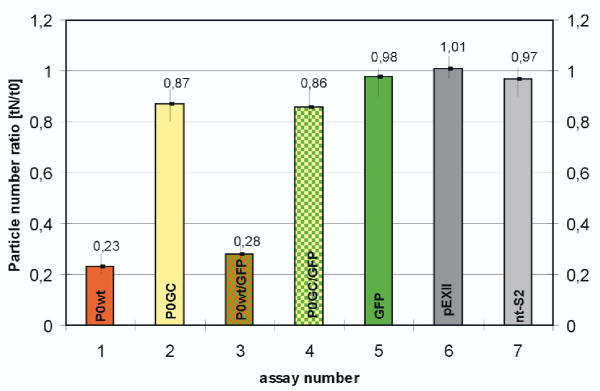
In contrast to the membrane-localized wild type P0-GFP the Ala221fs P0-GFP protein was detectable almost only in the cytoplasm of the cells, and a complete loss of adhesion function was observed.
The present study provides evidence that GFP is a versatile tool to trace in vivo effects of P0 and its mutations. Not only a loss of adhesion function as a result of the loss of the RSTK domain, but also altered intracellular trafficking indicated by a loss of membrane insertion are possible consequences of the Ala221fs mutation.
外周神经髓鞘的主要蛋白P0发生突变,会导致遗传性外周神经病1B型夏科-马里-图斯病(CMT1B)、德热里纳-索塔斯综合征(DSS)和先天性髓鞘形成低下(CH)。我们之前报道了一名DSS患者存在一个新生插入突变c.662_663GC(Ala221fs)。c.662_663GC插入导致移码突变Ala221fs,改变了C端氨基酸序列。与黏附相关的细胞内RSTK结构域被一个类似于钠钾ATP酶的序列取代。为了进一步阐明这名散发患者的分子疾病机制,我们通过定点诱变构建了野生型P0和c.662_663GC突变体表达盒,并将构建体转染到昆虫细胞(S2、High5)中。为了追踪活细胞中的效应,绿色荧光蛋白(GFP)已被添加到野生型和突变型P0蛋白的羧基末端。
与定位于膜上的野生型P0-GFP不同,Ala221fs P0-GFP蛋白几乎只能在细胞的细胞质中检测到,并且观察到黏附功能完全丧失。
本研究提供了证据表明,GFP是追踪P0及其突变在体内效应的通用工具。Ala221fs突变可能导致的后果不仅是由于RSTK结构域缺失导致黏附功能丧失,还包括膜插入缺失所表明的细胞内运输改变。