Goodstadt Leo, Ponting Chris P
Medical Research Council Functional Genetics Unit, University of Oxford, Department of Physiology, Anatomy, and Genetics, Oxford, United Kingdom.
PLoS Comput Biol. 2006 Sep 29;2(9):e133. doi: 10.1371/journal.pcbi.0020133.
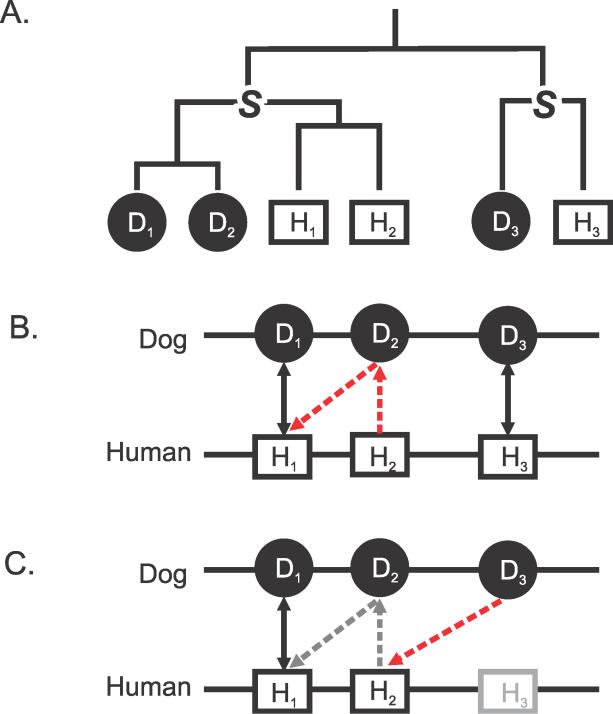
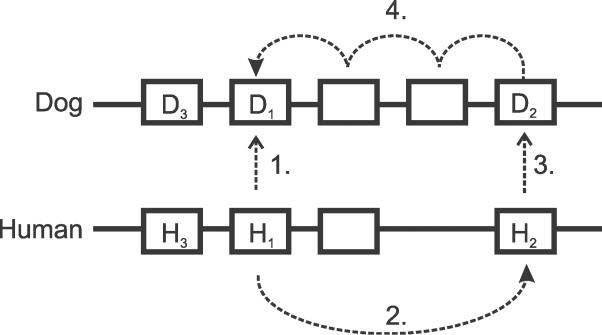
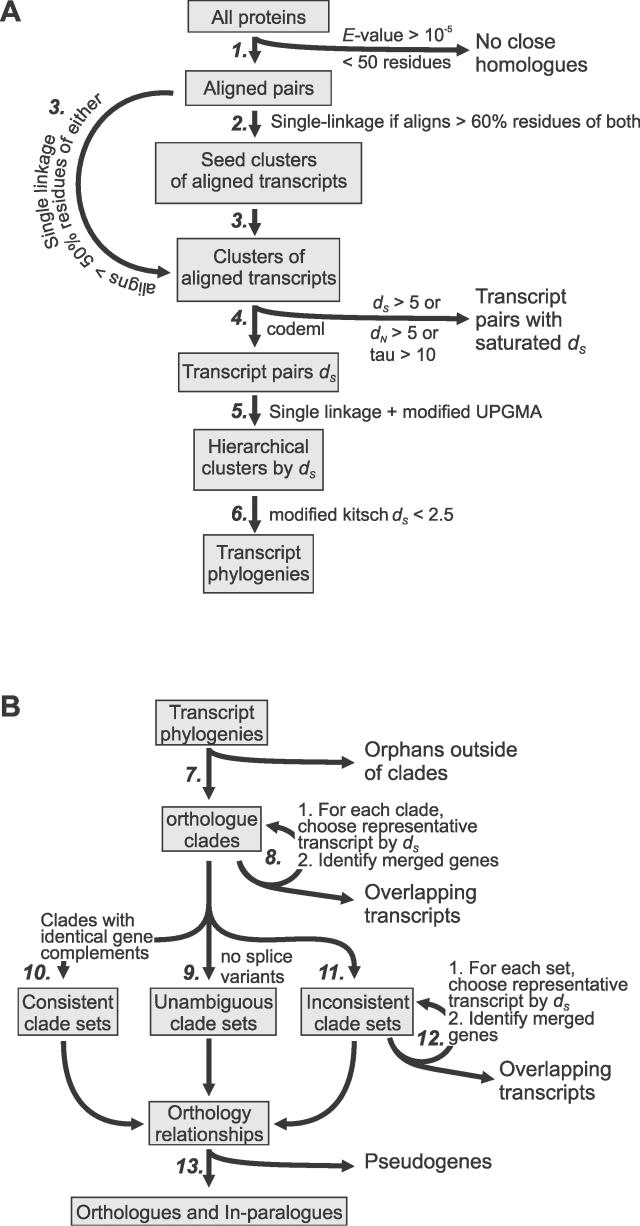
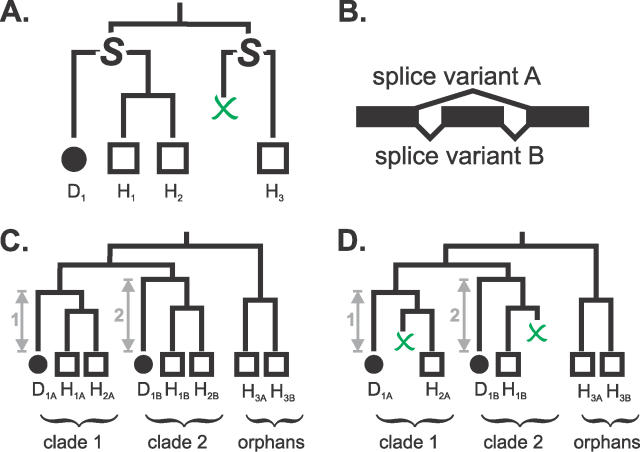
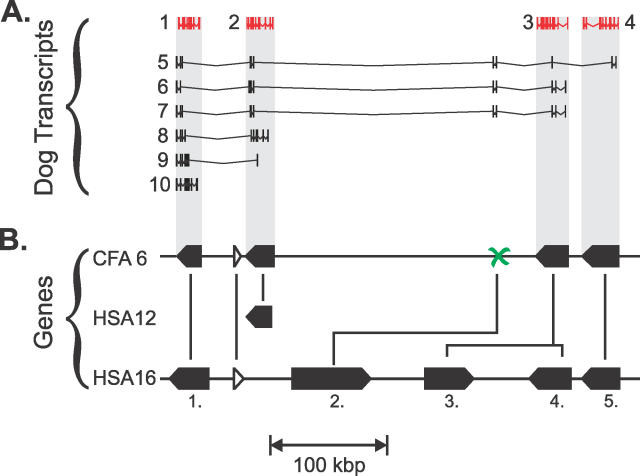
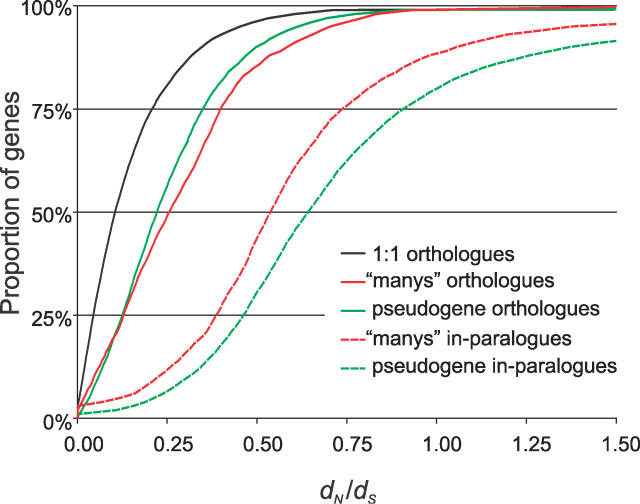
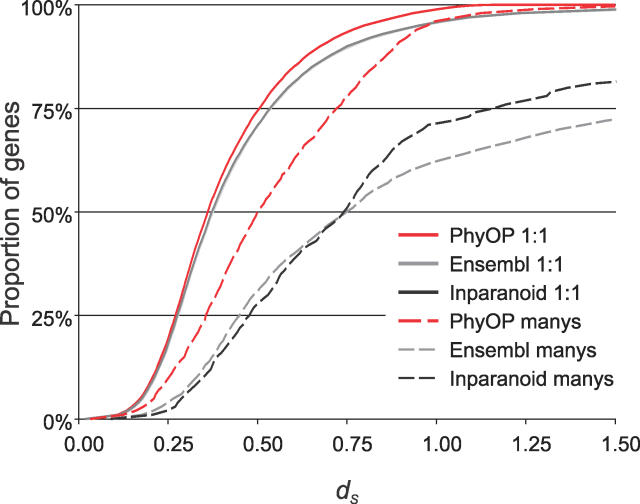
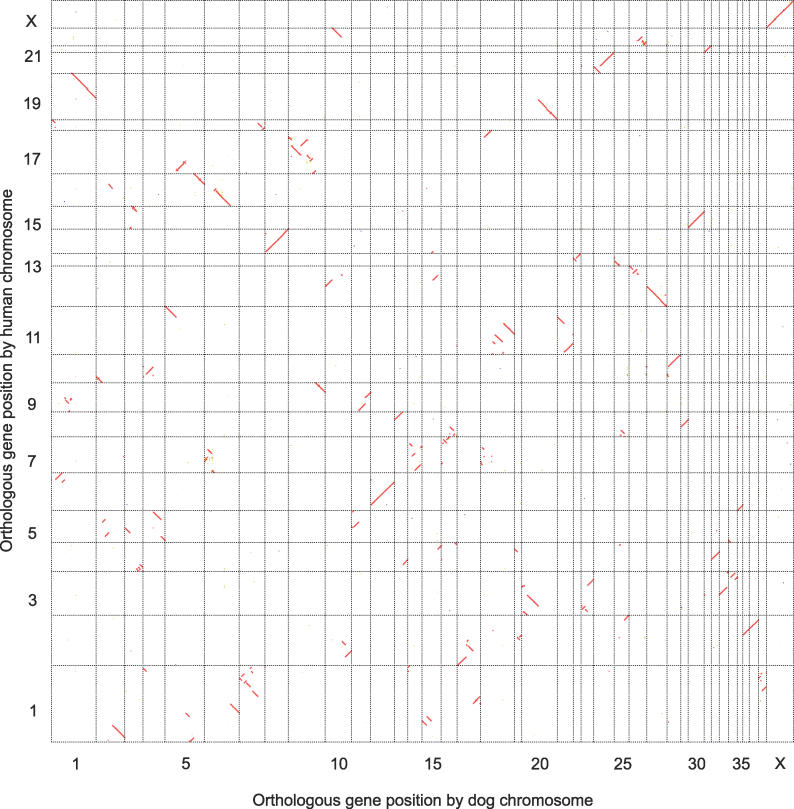
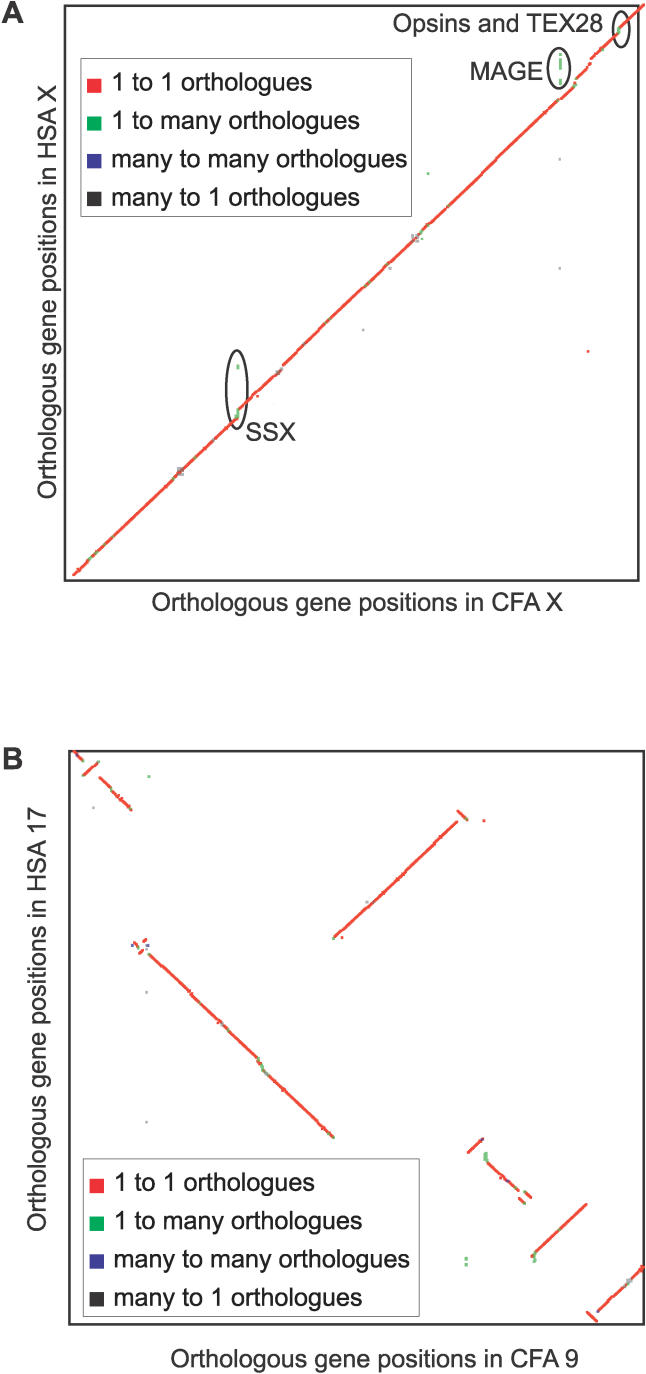
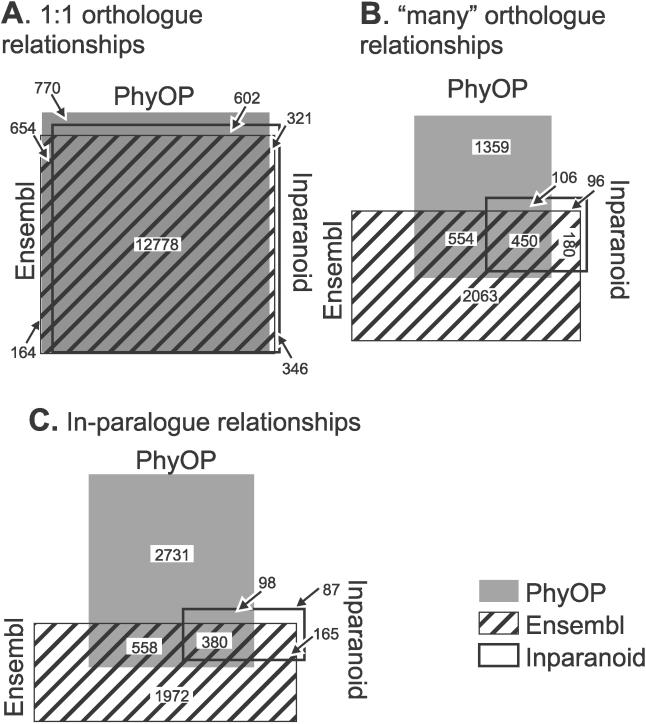
Accurate predictions of orthology and paralogy relationships are necessary to infer human molecular function from experiments in model organisms. Previous genome-scale approaches to predicting these relationships have been limited by their use of protein similarity and their failure to take into account multiple splicing events and gene prediction errors. We have developed PhyOP, a new phylogenetic orthology prediction pipeline based on synonymous rate estimates, which accurately predicts orthology and paralogy relationships for transcripts, genes, exons, or genomic segments between closely related genomes. We were able to identify orthologue relationships to human genes for 93% of all dog genes from Ensembl. Among 1:1 orthologues, the alignments covered a median of 97.4% of protein sequences, and 92% of orthologues shared essentially identical gene structures. PhyOP accurately recapitulated genomic maps of conserved synteny. Benchmarking against predictions from Ensembl and Inparanoid showed that PhyOP is more accurate, especially in its predictions of paralogy. Nearly half (46%) of PhyOP paralogy predictions are unique. Using PhyOP to investigate orthologues and paralogues in the human and dog genomes, we found that the human assembly contains 3-fold more gene duplications than the dog. Species-specific duplicate genes, or "in-paralogues," are generally shorter and have fewer exons than 1:1 orthologues, which is consistent with selective constraints and mutation biases based on the sizes of duplicated genes. In-paralogues have experienced elevated amino acid and synonymous nucleotide substitution rates. Duplicates possess similar biological functions for either the dog or human lineages. Having accounted for 2,954 likely pseudogenes and gene fragments, and after separating 346 erroneously merged genes, we estimated that the human genome encodes a minimum of 19,700 protein-coding genes, similar to the gene count of nematode worms. PhyOP is a fast and robust approach to orthology prediction that will be applicable to whole genomes from multiple closely related species. PhyOP will be particularly useful in predicting orthology for mammalian genomes that have been incompletely sequenced, and for large families of rapidly duplicating genes.
准确预测直系同源和旁系同源关系对于从模式生物实验中推断人类分子功能至关重要。先前用于预测这些关系的全基因组方法受到蛋白质相似性的限制,且未考虑多种剪接事件和基因预测错误。我们开发了PhyOP,一种基于同义速率估计的新的系统发育直系同源预测流程,它能准确预测密切相关基因组之间转录本、基因、外显子或基因组片段的直系同源和旁系同源关系。我们能够为Ensembl中93%的犬类基因鉴定出与人基因的直系同源关系。在1:1直系同源物中,比对覆盖蛋白质序列的中位数为97.4%,92%的直系同源物具有基本相同的基因结构。PhyOP准确地重现了保守共线性的基因组图谱。与Ensembl和Inparanoid的预测结果进行基准测试表明,PhyOP更准确,尤其是在旁系同源预测方面。PhyOP近一半(46%)的旁系同源预测是独特的。使用PhyOP研究人类和犬类基因组中的直系同源物和旁系同源物,我们发现人类基因组中的基因重复比犬类多3倍。物种特异性重复基因,即“内部旁系同源物”,通常比1:1直系同源物更短且外显子更少,这与基于重复基因大小的选择限制和突变偏差一致。内部旁系同源物经历了更高的氨基酸和同义核苷酸替换率。重复基因在犬类或人类谱系中具有相似的生物学功能。在考虑了2954个可能的假基因和基因片段,并分离了346个错误合并的基因后,我们估计人类基因组至少编码19700个蛋白质编码基因,与线虫的基因数量相似。PhyOP是一种快速且稳健的直系同源预测方法,将适用于多个密切相关物种的全基因组。PhyOP在预测未完全测序的哺乳动物基因组以及快速复制的基因大家族的直系同源关系方面将特别有用。