Marini Joan C, Forlino Antonella, Cabral Wayne A, Barnes Aileen M, San Antonio James D, Milgrom Sarah, Hyland James C, Körkkö Jarmo, Prockop Darwin J, De Paepe Anne, Coucke Paul, Symoens Sofie, Glorieux Francis H, Roughley Peter J, Lund Alan M, Kuurila-Svahn Kaija, Hartikka Heini, Cohn Daniel H, Krakow Deborah, Mottes Monica, Schwarze Ulrike, Chen Diana, Yang Kathleen, Kuslich Christine, Troendle James, Dalgleish Raymond, Byers Peter H
Bone and Extracellular Matrix Branch, National Institute of Child Health and Human Development, NIH, Bethesda, Maryland 20892, USA.
Hum Mutat. 2007 Mar;28(3):209-21. doi: 10.1002/humu.20429.
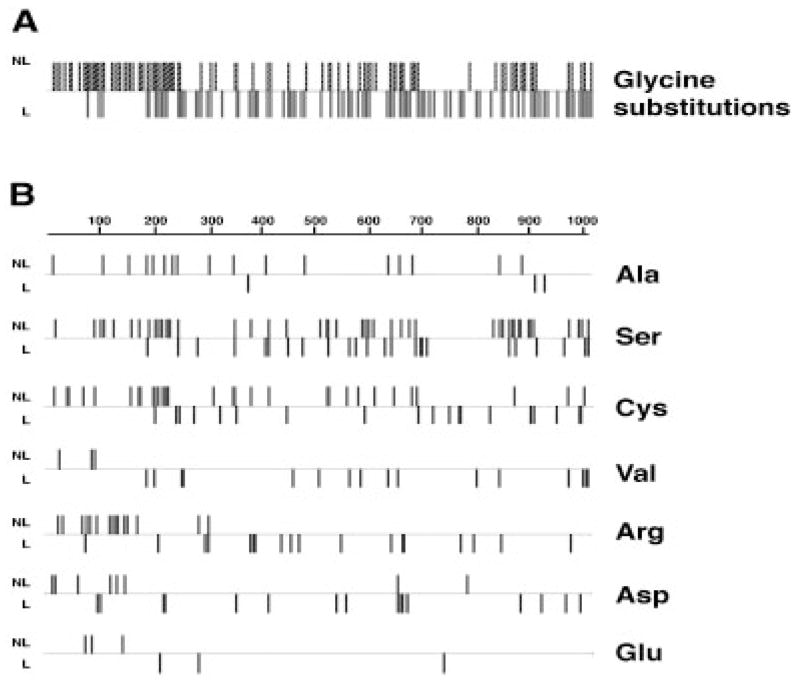
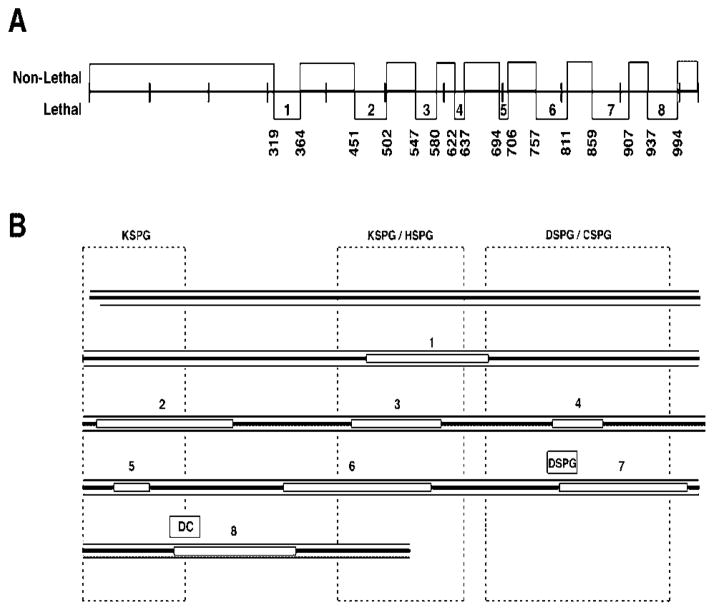
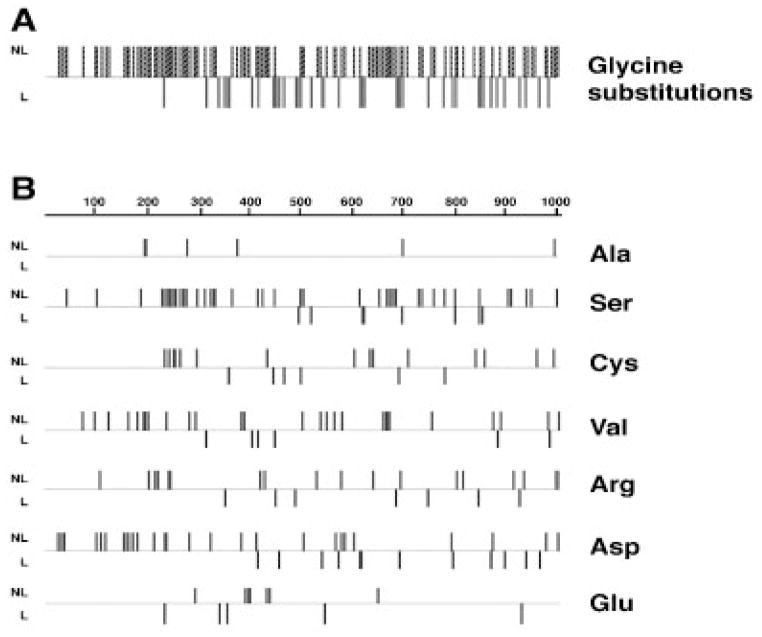
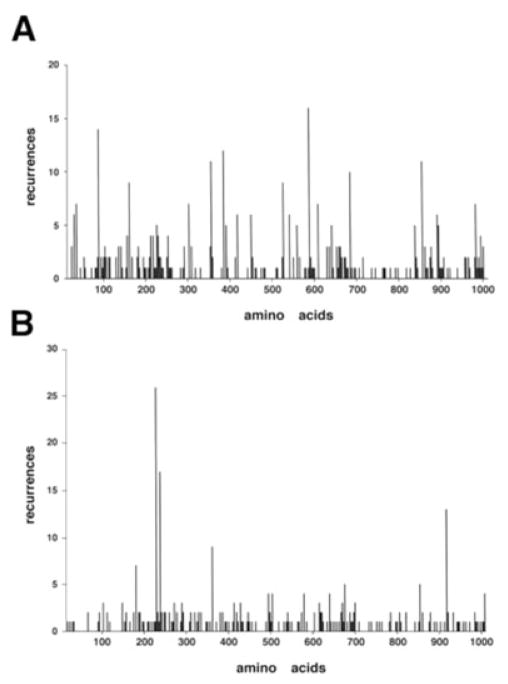
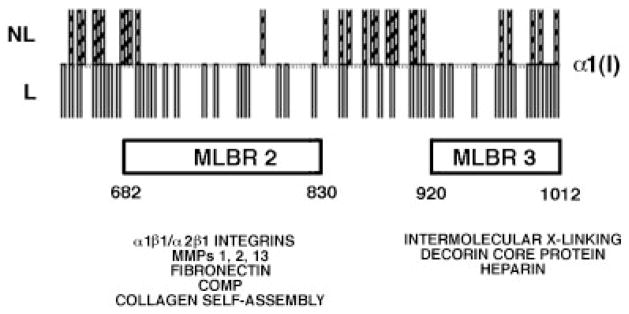
Osteogenesis imperfecta (OI) is a generalized disorder of connective tissue characterized by fragile bones and easy susceptibility to fracture. Most cases of OI are caused by mutations in type I collagen. We have identified and assembled structural mutations in type I collagen genes (COL1A1 and COL1A2, encoding the proalpha1(I) and proalpha2(I) chains, respectively) that result in OI. Quantitative defects causing type I OI were not included. Of these 832 independent mutations, 682 result in substitution for glycine residues in the triple helical domain of the encoded protein and 150 alter splice sites. Distinct genotype-phenotype relationships emerge for each chain. One-third of the mutations that result in glycine substitutions in alpha1(I) are lethal, especially when the substituting residues are charged or have a branched side chain. Substitutions in the first 200 residues are nonlethal and have variable outcome thereafter, unrelated to folding or helix stability domains. Two exclusively lethal regions (helix positions 691-823 and 910-964) align with major ligand binding regions (MLBRs), suggesting crucial interactions of collagen monomers or fibrils with integrins, matrix metalloproteinases (MMPs), fibronectin, and cartilage oligomeric matrix protein (COMP). Mutations in COL1A2 are predominantly nonlethal (80%). Lethal substitutions are located in eight regularly spaced clusters along the chain, supporting a regional model. The lethal regions align with proteoglycan binding sites along the fibril, suggesting a role in fibril-matrix interactions. Recurrences at the same site in alpha2(I) are generally concordant for outcome, unlike alpha1(I). Splice site mutations comprise 20% of helical mutations identified in OI patients, and may lead to exon skipping, intron inclusion, or the activation of cryptic splice sites. Splice site mutations in COL1A1 are rarely lethal; they often lead to frameshifts and the mild type I phenotype. In alpha2(I), lethal exon skipping events are located in the carboxyl half of the chain. Our data on genotype-phenotype relationships indicate that the two collagen chains play very different roles in matrix integrity and that phenotype depends on intracellular and extracellular events.
成骨不全症(OI)是一种全身性结缔组织疾病,其特征为骨骼脆弱且易骨折。大多数OI病例是由I型胶原蛋白的突变引起的。我们已经鉴定并汇总了I型胶原蛋白基因(COL1A1和COL1A2,分别编码前α1(I)链和前α2(I)链)中的结构突变,这些突变会导致OI。未包括导致I型OI的数量缺陷。在这832个独立突变中,682个导致编码蛋白三螺旋结构域中的甘氨酸残基被取代,150个改变了剪接位点。每条链都呈现出独特的基因型-表型关系。导致α1(I)中甘氨酸被取代的突变中有三分之一是致死性的,尤其是当取代残基带电或具有分支侧链时。前200个残基中的取代是非致死性的,此后结果各异,与折叠或螺旋稳定结构域无关。两个完全致死区域(螺旋位置691-823和910-964)与主要配体结合区域(MLBRs)对齐,这表明胶原蛋白单体或原纤维与整合素、基质金属蛋白酶(MMPs)、纤连蛋白和软骨寡聚基质蛋白(COMP)之间存在关键相互作用。COL1A2中的突变主要是非致死性的(80%)。致死性取代位于沿链的八个规则间隔的簇中,支持区域模型。致死区域与沿原纤维的蛋白聚糖结合位点对齐,表明在原纤维-基质相互作用中起作用。与α1(I)不同,α2(I)中同一位置的复发通常在结果上是一致的。剪接位点突变占OI患者中鉴定出的螺旋突变的20%,可能导致外显子跳跃、内含子包含或隐蔽剪接位点的激活。COL1A1中的剪接位点突变很少是致死性的;它们通常会导致移码和轻度I型表型。在α2(I)中,致死性外显子跳跃事件位于链的羧基末端一半。我们关于基因型-表型关系的数据表明,两条胶原蛋白链在基质完整性中发挥着非常不同的作用,并且表型取决于细胞内和细胞外事件。