Muscarella Donna E, Bloom Stephen E
Department of Microbiology and Immunology, Cornell University, Ithaca, NY 14853, USA.
Toxicol Appl Pharmacol. 2008 Apr 1;228(1):93-104. doi: 10.1016/j.taap.2007.11.032. Epub 2007 Dec 14.
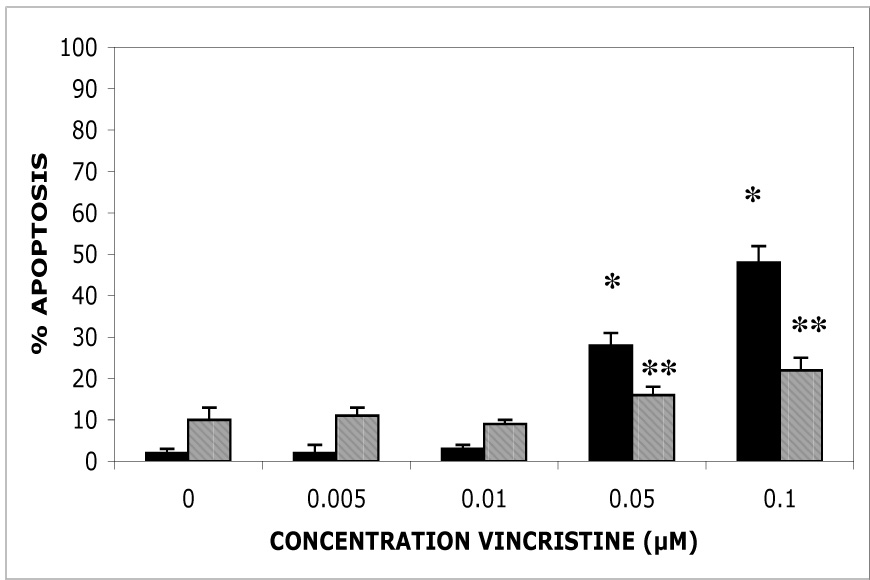
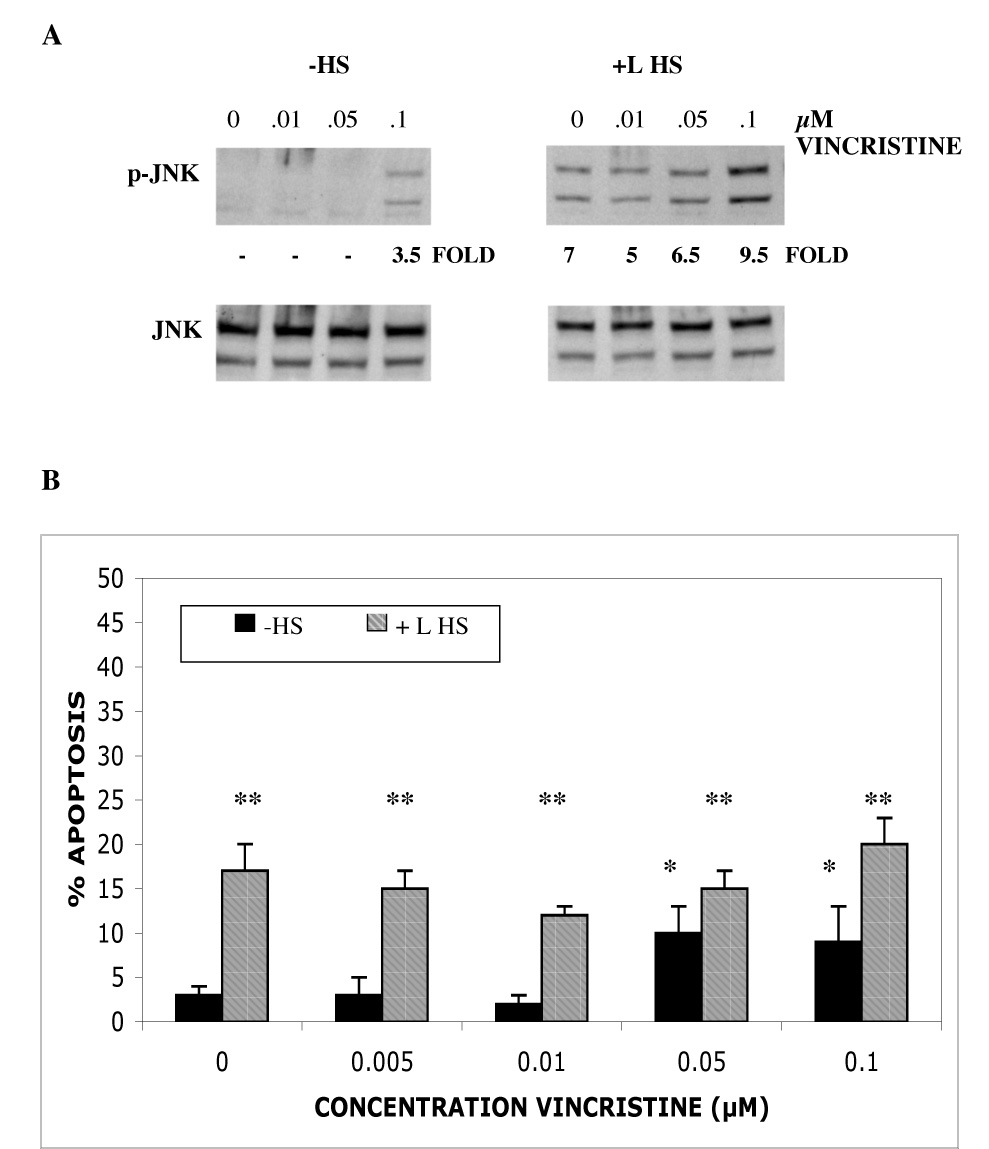
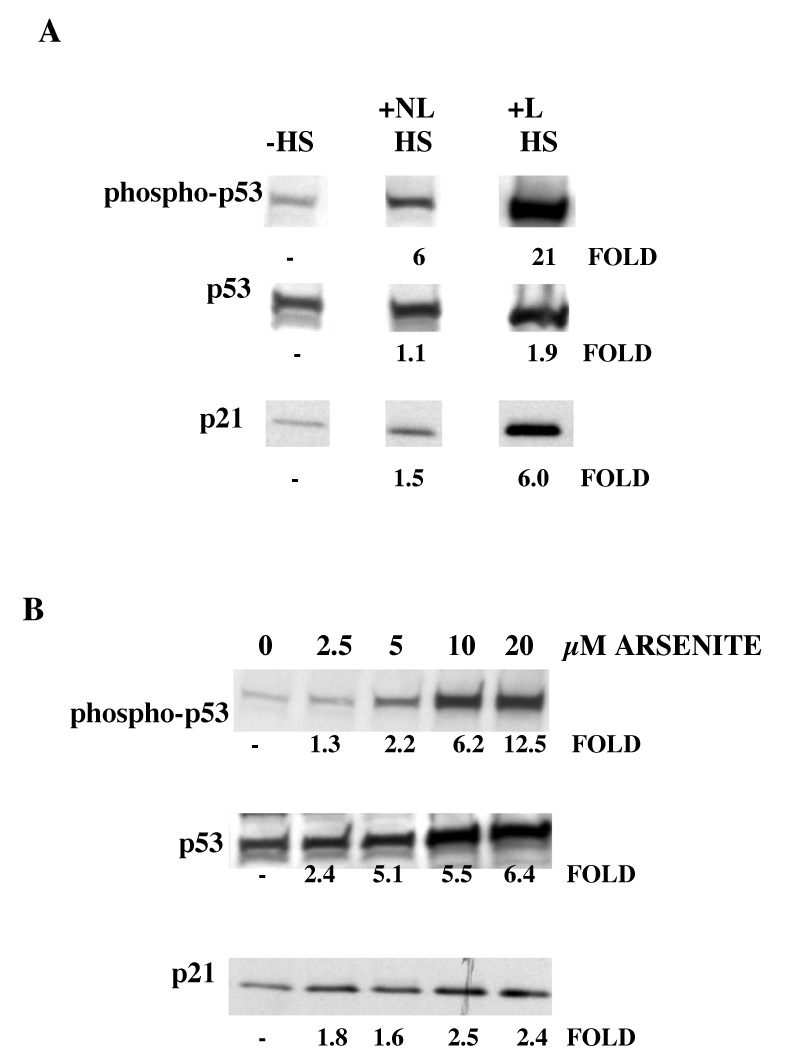
The c-Jun N-terminal kinase (JNK) pathway can play paradoxical roles as either a pro-survival or a pro-cell death pathway depending on type of stress and cell type. The goal of the present study was to determine the role of JNK pathway signaling for regulating B-cell apoptosis in two important but contrasting situations--global proteotoxic damage, induced by arsenite and hyperthermia, versus specific microtubule inhibition, induced by the anti-cancer drug vincristine, using the EW36 B-cell line. This cell line over-expresses the Bcl-2 protein and is a useful model to identify treatments that can overcome multi-drug resistance in lymphoid cells. Exposure of EW36 B-cells to arsenite or lethal hyperthermia resulted in activation of the JNK pathway and induction of apoptosis. However, pharmacological inhibition of the JNK pathway did not inhibit apoptosis, indicating that JNK pathway activation is not required for apoptosis induction by these treatments. In contrast, vincristine treatment of EW36 B-cells resulted in JNK activation and apoptosis that was suppressed by JNK inhibition. A critical difference between the two types of stress treatments was that only vincristine-induced JNK activation resulted in phosphorylation of Bcl-2 at threonine-56, a modification that can block its anti-apoptotic function. Importantly, Bcl-2 phosphorylation was attenuated by JNK inhibition implicating JNK as the upstream kinase. Furthermore, arsenite and hyperthermia treatments activated a p53/p21 pathway associated with apoptosis induction, whereas vincristine did not activate this pathway. These results reveal two stress-activated pathways, one JNK-dependent and another JNK-independent, either of which can bypass Bcl-2 mediated resistance, resulting in cell death.
c-Jun氨基末端激酶(JNK)通路可发挥矛盾的作用,根据应激类型和细胞类型,它既可以是促生存通路,也可以是促细胞死亡通路。本研究的目的是利用EW36 B细胞系,确定JNK通路信号在两种重要但相反的情况下对调节B细胞凋亡的作用——由亚砷酸盐和热疗诱导的全身性蛋白毒性损伤,与由抗癌药物长春新碱诱导的特异性微管抑制。该细胞系过表达Bcl-2蛋白,是一种用于鉴定可克服淋巴细胞多药耐药性的治疗方法的有用模型。将EW36 B细胞暴露于亚砷酸盐或致死性热疗会导致JNK通路激活和凋亡诱导。然而,JNK通路的药理学抑制并未抑制凋亡,这表明这些处理诱导凋亡并不需要JNK通路激活。相比之下,长春新碱处理EW36 B细胞会导致JNK激活和凋亡,而这种凋亡会被JNK抑制所抑制。两种应激处理类型之间的一个关键差异在于,只有长春新碱诱导的JNK激活会导致Bcl-2在苏氨酸56处磷酸化,这种修饰可阻断其抗凋亡功能。重要的是,JNK抑制可减弱Bcl-2磷酸化,这表明JNK是上游激酶。此外,亚砷酸盐和热疗处理激活了与凋亡诱导相关的p53/p21通路,而长春新碱未激活该通路。这些结果揭示了两条应激激活通路,一条依赖JNK,另一条不依赖JNK,其中任何一条都可以绕过Bcl-2介导的抗性,导致细胞死亡。