Bean James, Riely Gregory J, Balak Marissa, Marks Jenifer L, Ladanyi Marc, Miller Vincent A, Pao William
Human Oncology and Pathogenesis Program, Memorial Sloan-Kettering Cancer Center, New York, NY 10021, USA.
Clin Cancer Res. 2008 Nov 15;14(22):7519-25. doi: 10.1158/1078-0432.CCR-08-0151.
Somatic mutations in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) gene are associated with sensitivity of lung adenocarcinomas to the EGFR tyrosine kinase inhibitors, gefitinib and erlotinib. Acquired drug resistance is frequently associated with a secondary somatic mutation that leads to the substitution of methionine for threonine at position 790 (T790M). We aimed to identify additional second-site alterations associated with acquired resistance.
Tumor samples were obtained from 48 patients with acquired resistance. Tumor cell DNA was analyzed for EGFR kinase domain mutations. Molecular analyses were then done to characterize the biological properties of a novel mutant EGFR allele.
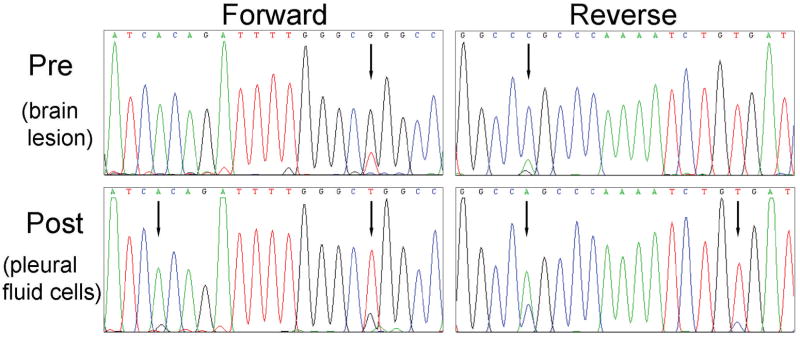
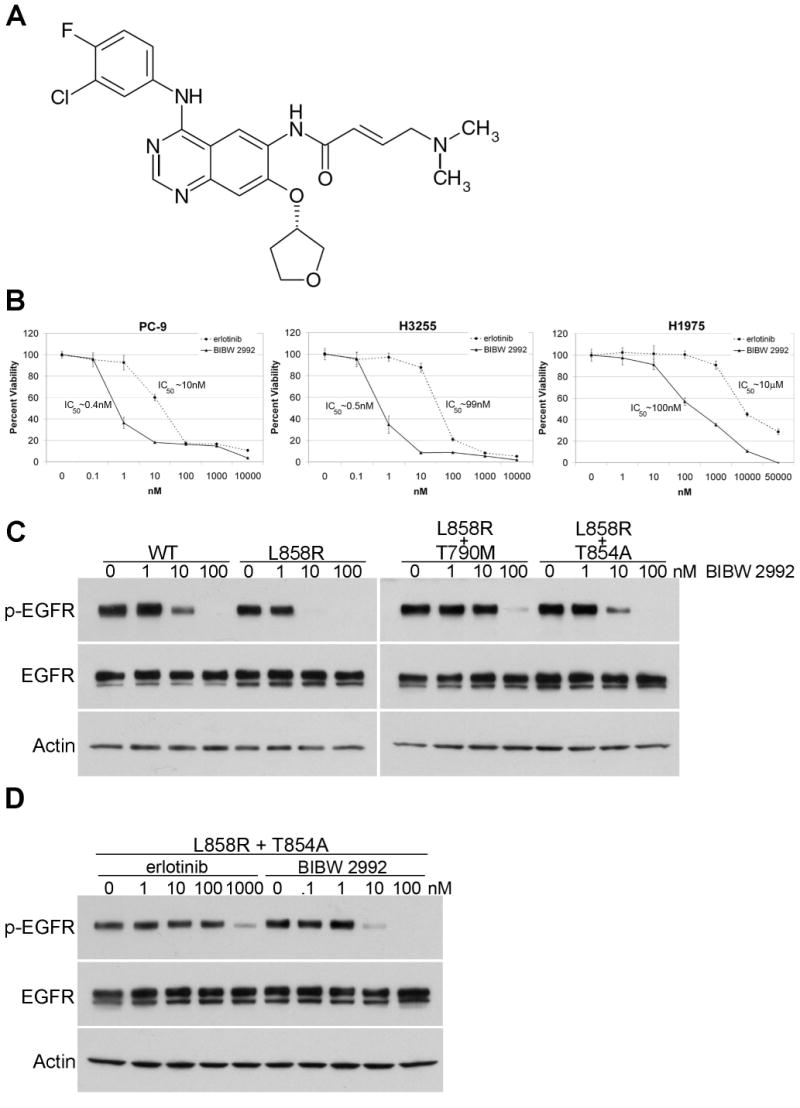
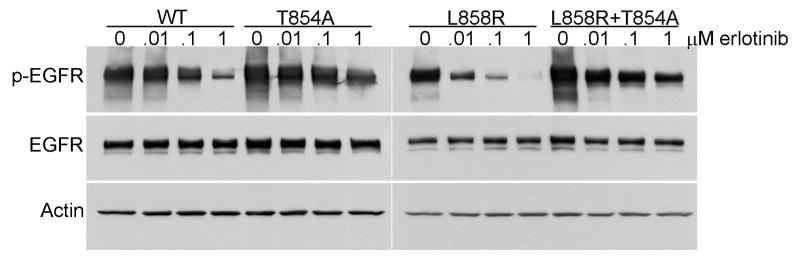
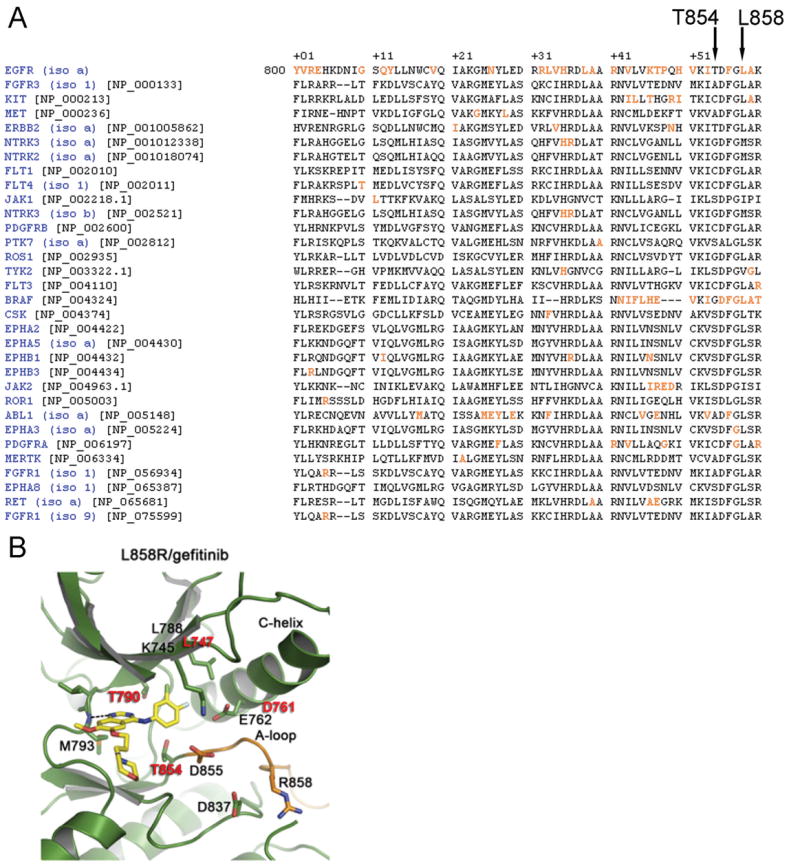
A previously unreported mutation in exon 21 of EGFR, which leads to substitution of alanine for threonine at position 854 (T854A), was identified in one patient with a drug-sensitive EGFR L858R-mutant lung adenocarcinoma after long-term treatment with tyrosine kinase inhibitors. The T854A mutation was not detected in a pretreatment tumor sample. The crystal structure analyses of EGFR suggest that the T854 side chain is within contact distance of gefitinib and erlotinib. Surrogate kinase assays show that the EGFR T854A mutation abrogates the inhibition of tyrosine phosphorylation by erlotinib. Such resistance seems to be overcome by a new irreversible dual EGFR/HER2 inhibitor, BIBW 2992.
The T854A mutation is the second reported second-site acquired resistance mutation that is within contact distance of gefitinib and erlotinib. These data suggest that acquired resistance to ATP-mimetic EGFR kinase inhibitors may often be associated with amino acid substitutions that alter drug contact residues in the EGFR ATP-binding pocket.
表皮生长因子受体(EGFR)基因酪氨酸激酶结构域中的体细胞突变与肺腺癌对EGFR酪氨酸激酶抑制剂吉非替尼和厄洛替尼的敏感性相关。获得性耐药通常与导致第790位(T790M)苏氨酸被甲硫氨酸替代的继发性体细胞突变有关。我们旨在识别与获得性耐药相关的其他第二位点改变。
从48例获得性耐药患者中获取肿瘤样本。分析肿瘤细胞DNA中的EGFR激酶结构域突变。然后进行分子分析以表征一种新型突变EGFR等位基因的生物学特性。
在一名对药物敏感的EGFR L858R突变型肺腺癌患者经酪氨酸激酶抑制剂长期治疗后,在其EGFR第21外显子中发现了一个先前未报道的突变,该突变导致第854位(T854A)苏氨酸被丙氨酸替代。在预处理的肿瘤样本中未检测到T854A突变。EGFR的晶体结构分析表明,T854侧链与吉非替尼和厄洛替尼处于接触距离内。替代激酶试验表明,EGFR T854A突变消除了厄洛替尼对酪氨酸磷酸化的抑制作用。一种新的不可逆双EGFR/HER2抑制剂BIBW 2992似乎可以克服这种耐药性。
T854A突变是第二个报道的与吉非替尼和厄洛替尼处于接触距离内的第二位点获得性耐药突变。这些数据表明,对ATP模拟物EGFR激酶抑制剂的获得性耐药可能常与改变EGFR ATP结合口袋中药物接触残基的氨基酸替代有关。