Dowling James J, Vreede Andrew P, Low Sean E, Gibbs Elizabeth M, Kuwada John Y, Bonnemann Carsten G, Feldman Eva L
Department of Pediatrics, University of Michigan Medical Center, Ann Arbor, Michigan, USA.
PLoS Genet. 2009 Feb;5(2):e1000372. doi: 10.1371/journal.pgen.1000372. Epub 2009 Feb 6.
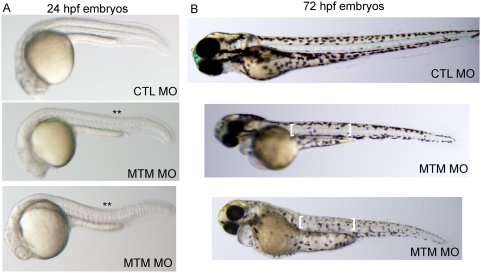
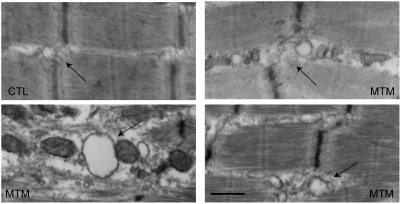
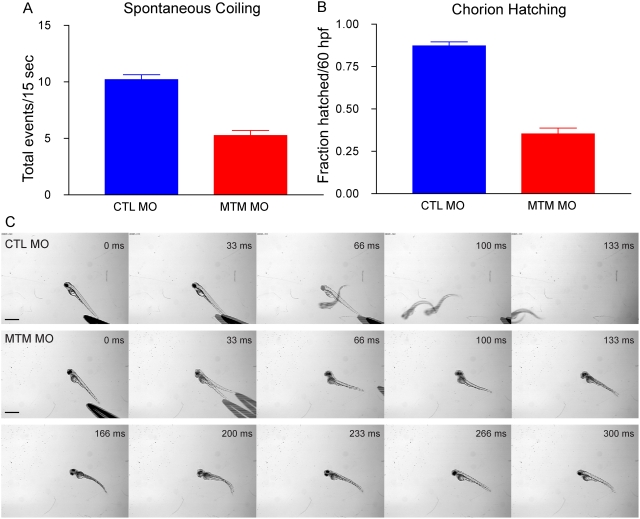
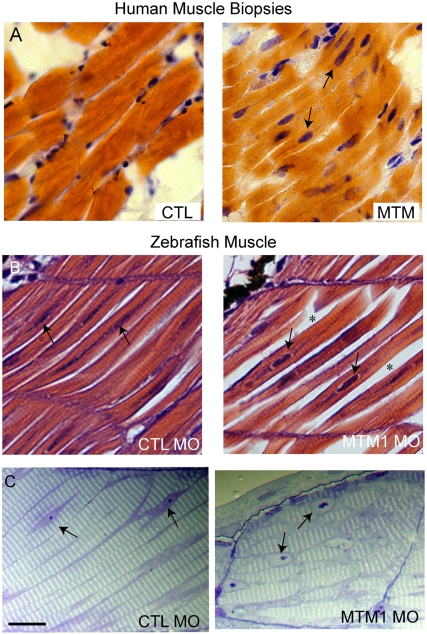
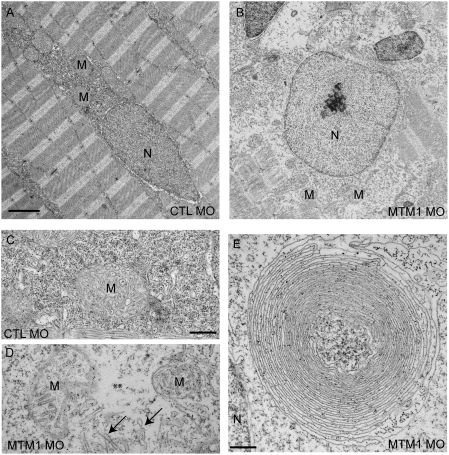
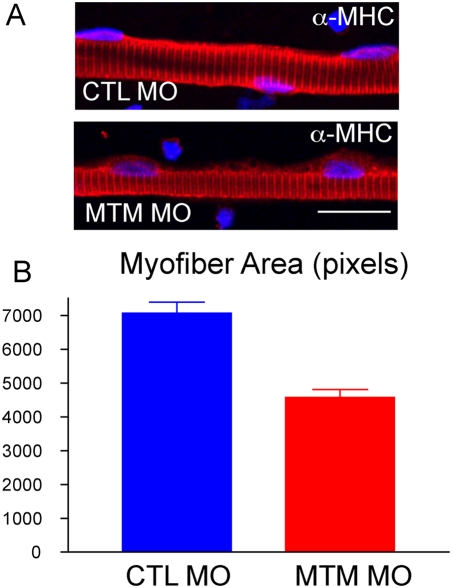
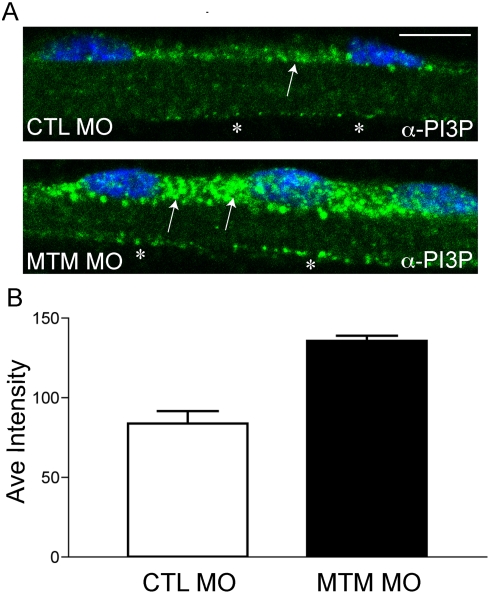
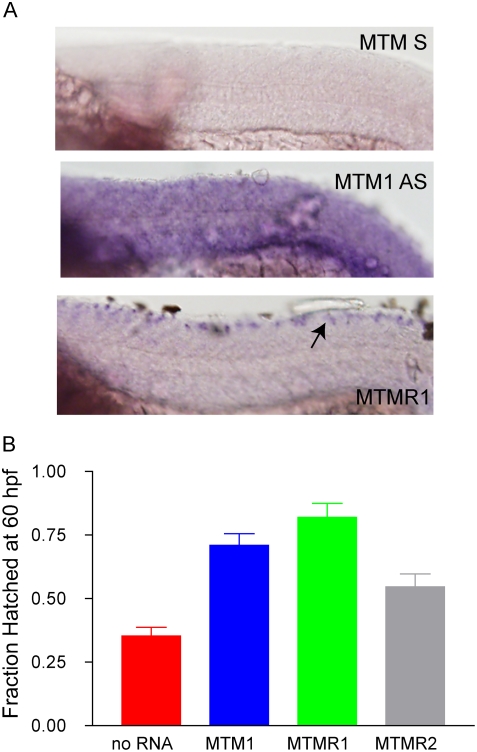
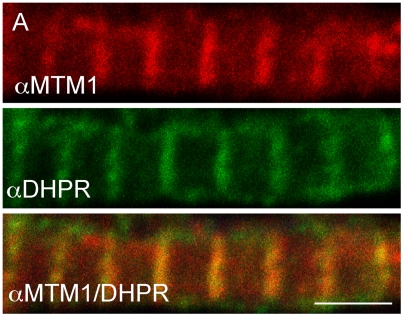
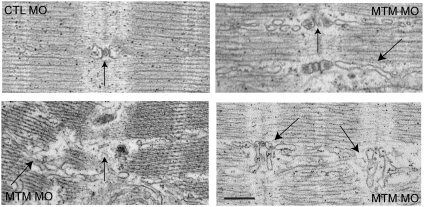
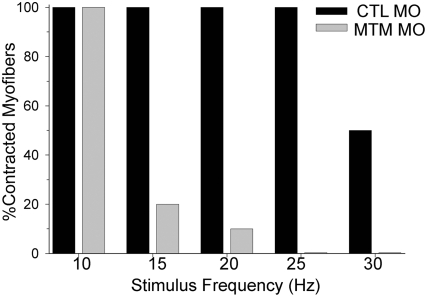
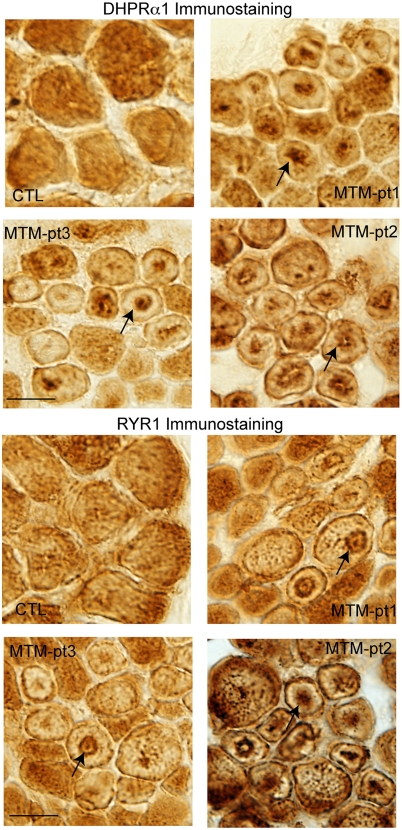
Myotubularin is a lipid phosphatase implicated in endosomal trafficking in vitro, but with an unknown function in vivo. Mutations in myotubularin cause myotubular myopathy, a devastating congenital myopathy with unclear pathogenesis and no current therapies. Myotubular myopathy was the first described of a growing list of conditions caused by mutations in proteins implicated in membrane trafficking. To advance the understanding of myotubularin function and disease pathogenesis, we have created a zebrafish model of myotubular myopathy using morpholino antisense technology. Zebrafish with reduced levels of myotubularin have significantly impaired motor function and obvious histopathologic changes in their muscle. These changes include abnormally shaped and positioned nuclei and myofiber hypotrophy. These findings are consistent with those observed in the human disease. We demonstrate for the first time that myotubularin functions to regulate PI3P levels in a vertebrate in vivo, and that homologous myotubularin-related proteins can functionally compensate for the loss of myotubularin. Finally, we identify abnormalities in the tubulo-reticular network in muscle from myotubularin zebrafish morphants and correlate these changes with abnormalities in T-tubule organization in biopsies from patients with myotubular myopathy. In all, we have generated a new model of myotubular myopathy and employed this model to uncover a novel function for myotubularin and a new pathomechanism for the human disease that may explain the weakness associated with the condition (defective excitation-contraction coupling). In addition, our findings of tubuloreticular abnormalities and defective excitation-contraction coupling mechanistically link myotubular myopathy with several other inherited muscle diseases, most notably those due to ryanodine receptor mutations. Based on our findings, we speculate that congenital myopathies, usually considered entities with similar clinical features but very disparate pathomechanisms, may at their root be disorders of calcium homeostasis.
肌管素是一种脂质磷酸酶,在体外参与内体运输,但在体内功能未知。肌管素突变会导致肌管性肌病,这是一种严重的先天性肌病,发病机制不明,目前尚无有效治疗方法。肌管性肌病是首批被描述的由参与膜运输的蛋白质突变引起的一系列病症之一。为了加深对肌管素功能和疾病发病机制的理解,我们利用吗啉代反义技术创建了肌管性肌病的斑马鱼模型。肌管素水平降低的斑马鱼运动功能显著受损,肌肉出现明显的组织病理学变化。这些变化包括细胞核形状和位置异常以及肌纤维萎缩。这些发现与人类疾病中观察到的一致。我们首次证明,肌管素在脊椎动物体内发挥调节磷脂酰肌醇3-磷酸(PI3P)水平的作用,并且同源的肌管素相关蛋白可以在功能上补偿肌管素的缺失。最后,我们在肌管素斑马鱼形态突变体的肌肉中发现了管状网状网络异常,并将这些变化与肌管性肌病患者活检中T小管组织异常相关联。总之,我们建立了一种新的肌管性肌病模型,并利用该模型揭示了肌管素的新功能以及人类疾病的新发病机制,这可能解释了与该病症相关的肌无力(兴奋-收缩偶联缺陷)。此外,我们关于管状网状异常和兴奋-收缩偶联缺陷的发现从机制上将肌管性肌病与其他几种遗传性肌肉疾病联系起来,最显著的是那些由雷诺丁受体突变引起的疾病。基于我们的发现,我们推测先天性肌病通常被认为是具有相似临床特征但发病机制截然不同的实体,其根源可能是钙稳态紊乱。