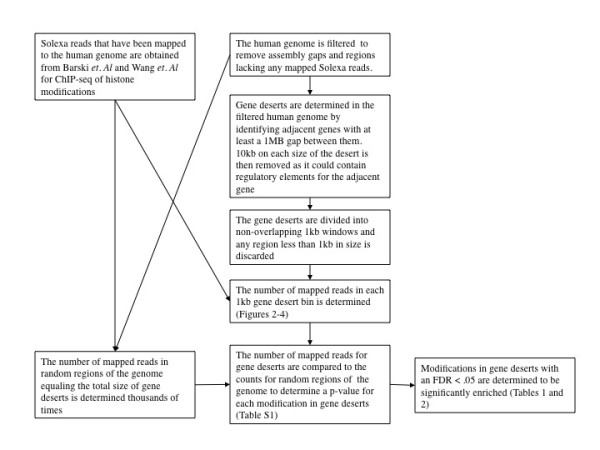
Rosenfeld Jeffrey A, Wang Zhibin, Schones Dustin E, Zhao Keji, DeSalle Rob, Zhang Michael Q
Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
BMC Genomics. 2009 Mar 31;10:143. doi: 10.1186/1471-2164-10-143.
Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) has recently been used to identify the modification patterns for the methylation and acetylation of many different histone tails in genes and enhancers.
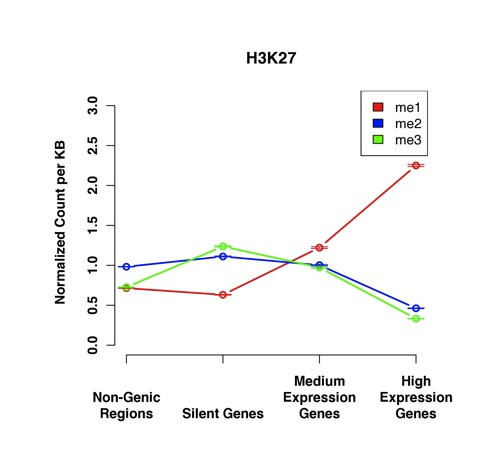
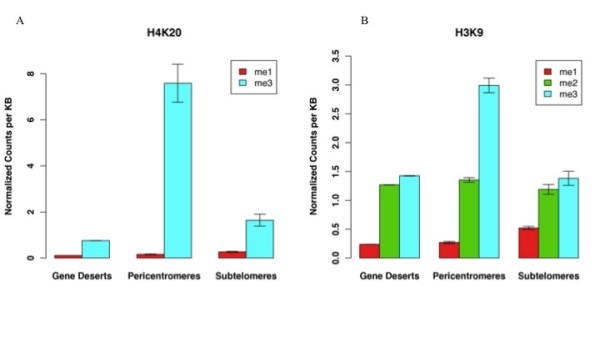
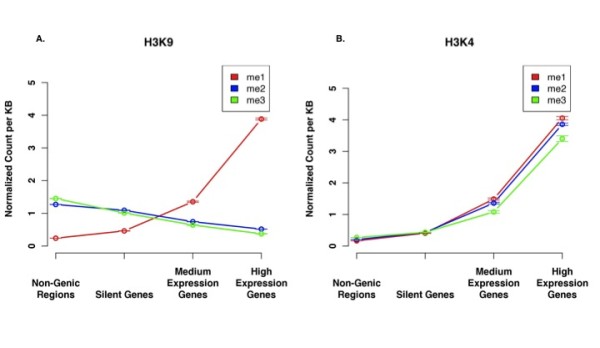
We have extended the analysis of histone modifications to gene deserts, pericentromeres and subtelomeres. Using data from human CD4+ T cells, we have found that each of these non-genic regions has a particular profile of histone modifications that distinguish it from the other non-coding regions. Different methylation states of H4K20, H3K9 and H3K27 were found to be enriched in each region relative to the other regions. These findings indicate that non-genic regions of the genome are variable with respect to histone modification patterns, rather than being monolithic. We furthermore used consensus sequences for unassembled centromeres and telomeres to identify the significant histone modifications in these regions. Finally, we compared the modification patterns in non-genic regions to those at silent genes and genes with higher levels of expression. For all tested methylations with the exception of H3K27me3, the enrichment level of each modification state for silent genes is between that of non-genic regions and expressed genes. For H3K27me3, the highest levels are found in silent genes.
In addition to the histone modification pattern difference between euchromatin and heterochromatin regions, as is illustrated by the enrichment of H3K9me2/3 in non-genic regions while H3K9me1 is enriched at active genes; the chromatin modifications within non-genic (heterochromatin-like) regions (e.g. subtelomeres, pericentromeres and gene deserts) are also quite different.
染色质免疫沉淀结合高通量测序(ChIP-seq)最近已被用于识别基因和增强子中许多不同组蛋白尾部的甲基化和乙酰化修饰模式。
我们将组蛋白修饰的分析扩展到基因沙漠、着丝粒周围区域和亚端粒区域。利用来自人类CD4 + T细胞的数据,我们发现这些非基因区域中的每一个都有特定的组蛋白修饰谱,使其与其他非编码区域区分开来。相对于其他区域,发现H4K20、H3K9和H3K27的不同甲基化状态在每个区域中富集。这些发现表明,基因组的非基因区域在组蛋白修饰模式方面是可变的,而不是单一的。我们还使用了未组装的着丝粒和端粒的共有序列来识别这些区域中显著的组蛋白修饰。最后,我们将非基因区域的修饰模式与沉默基因和高表达基因的修饰模式进行了比较。除H3K27me3外,对于所有测试的甲基化,沉默基因的每种修饰状态的富集水平介于非基因区域和表达基因之间。对于H3K27me3,在沉默基因中发现的水平最高。
除了常染色质和异染色质区域之间的组蛋白修饰模式差异,如非基因区域中H3K9me2 / 3的富集而活性基因中H3K9me1的富集所表明的那样;非基因(类异染色质)区域(例如亚端粒、着丝粒周围区域和基因沙漠)内的染色质修饰也有很大不同。