Division of Molecular Pharmaceutics, Center for Nanotechnology in Drug Delivery, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
J Biomed Nanotechnol. 2009 Apr;5(2):151-61. doi: 10.1166/jbn.2009.1021.
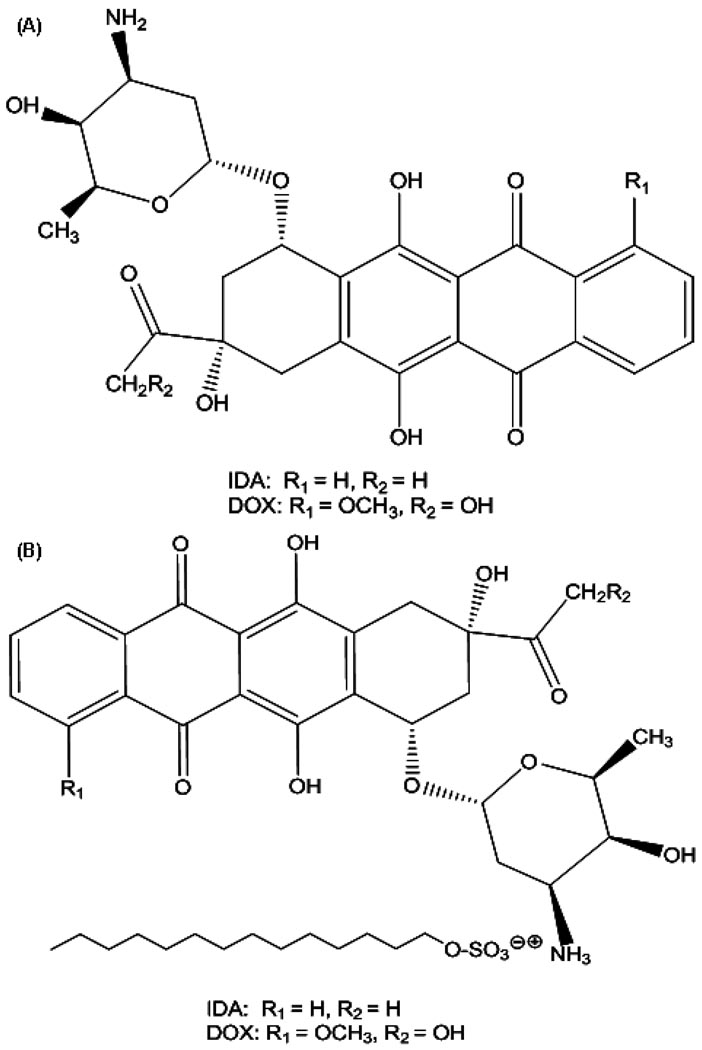
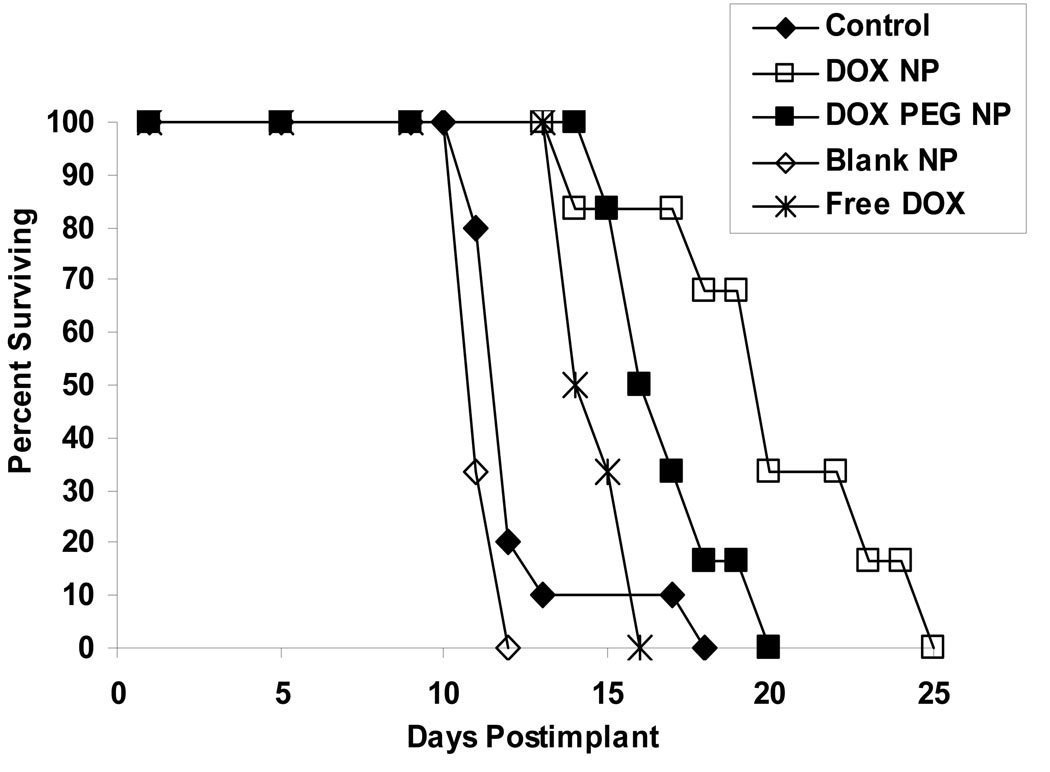
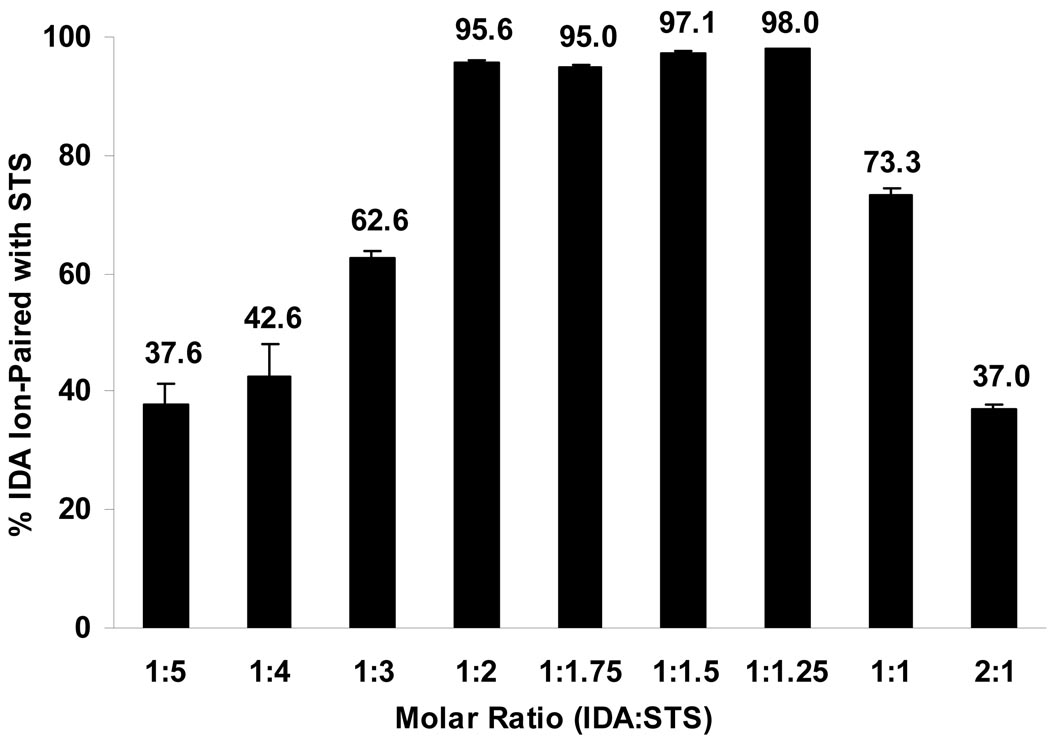
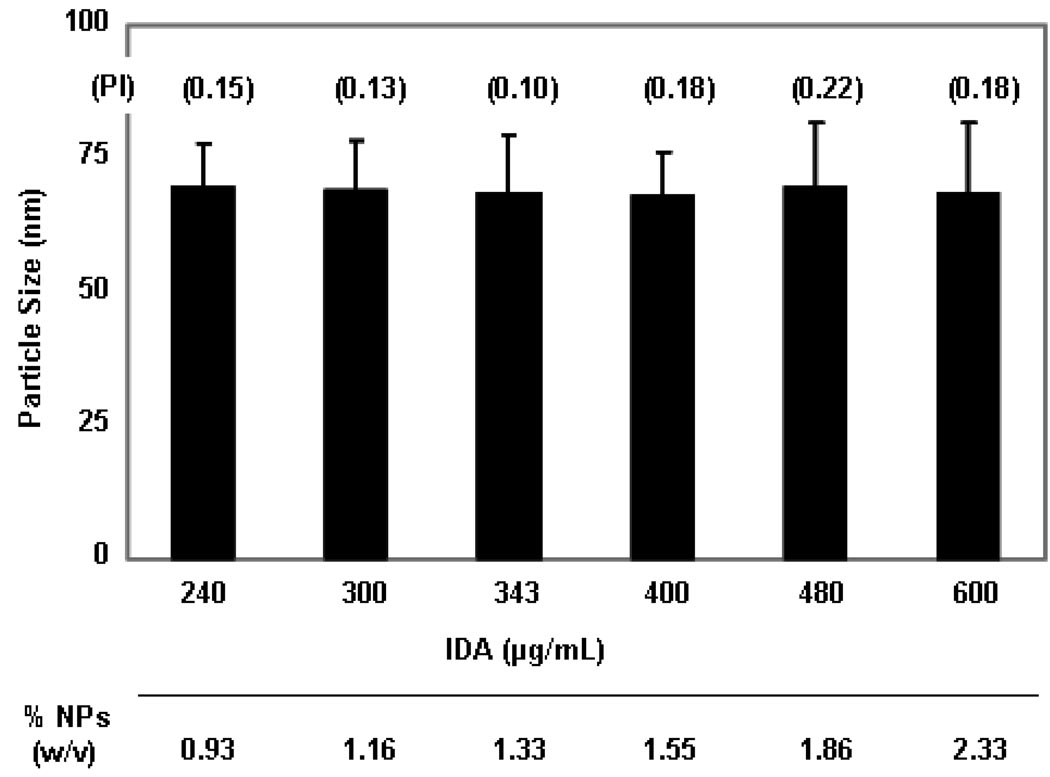
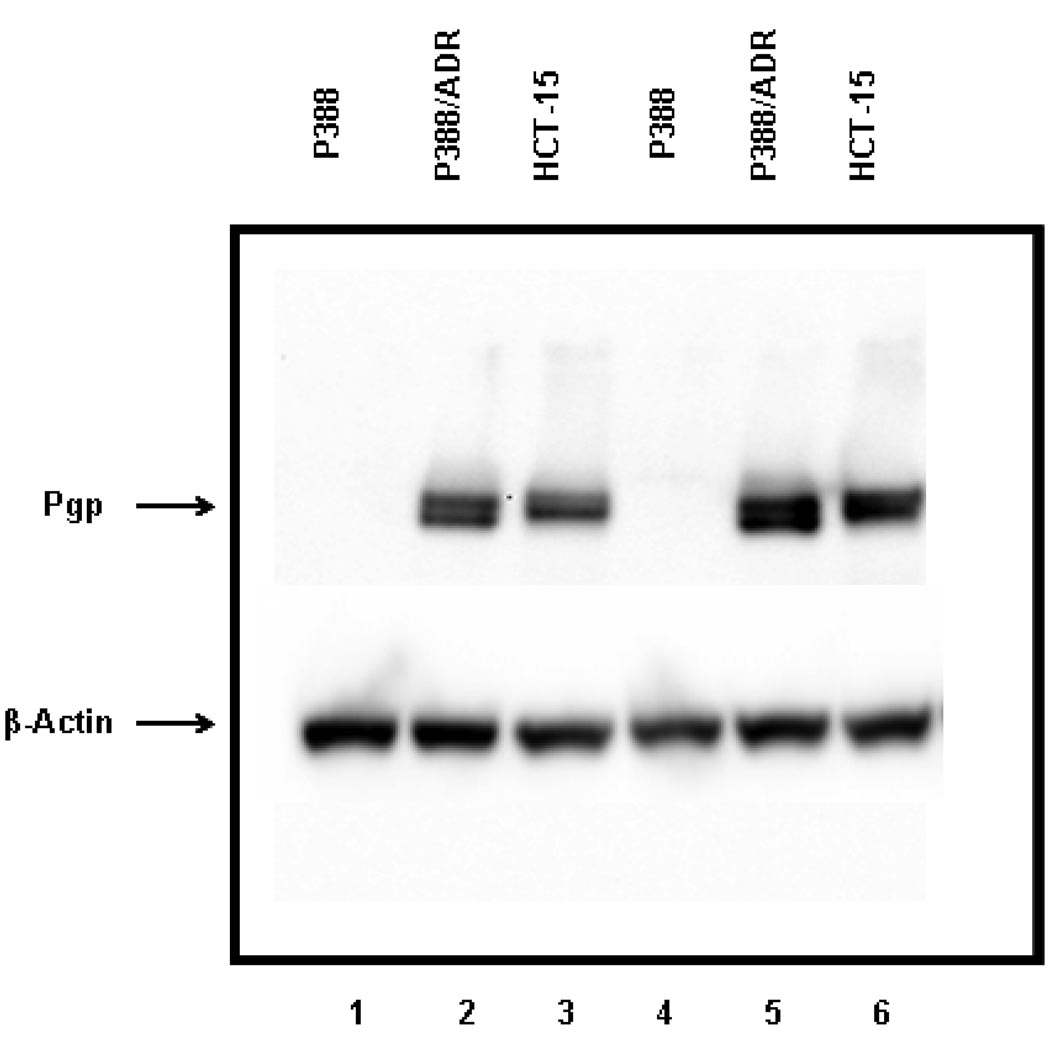
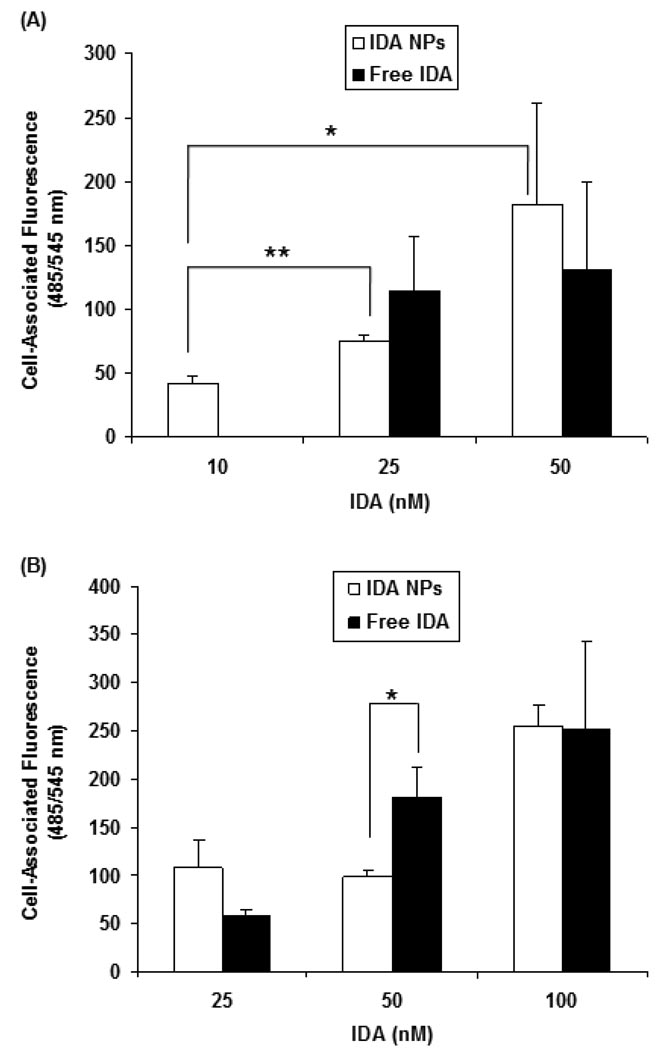
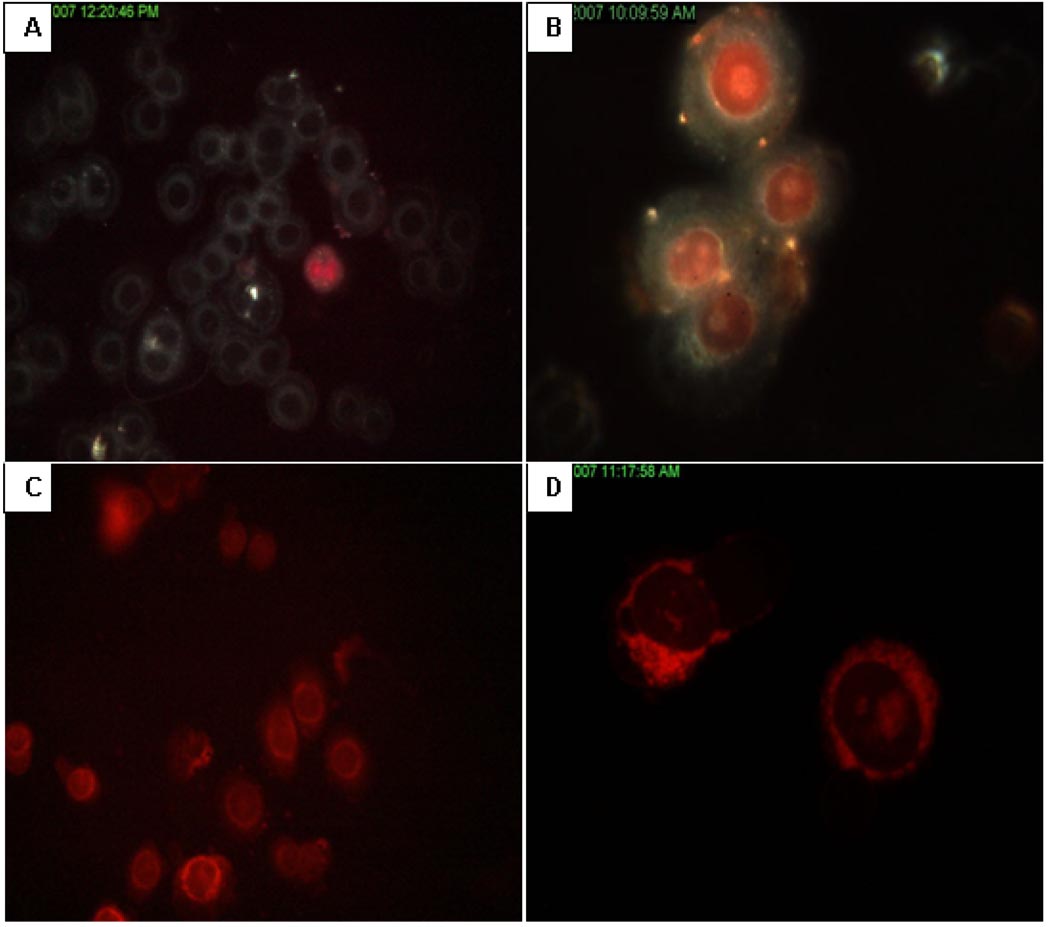
The objectives of these studies were to investigate and compare solid lipid nanoparticles (SLNs) of two anthracyclines, idarubicin (IDA) and doxorubicin (DOX), against Pgp-mediated multiple drug resistance (MDR) in-vitro and in-vivo using different human and murine cancer cell models. IDA and DOX SLNs were developed from warm microemulsion precursors comprising emulsifying wax as the oil phase, and polyoxyl 20-stearyl ether (Brij 78) and D-alpha-tocopheryl polyethylene glycol succinate (Vitamin E TPGS) as the surfactants. Anionic ion-pairing agents, sodium taurodeoxycholate (STDC) and sodium tetradecyl sulfate (STS), were used to neutralize the charges of the cationic anthracyclines and enhance entrapment of the drugs in the SLN. The in-vitro cytotoxicity results showed that the IC50 value of DOX NPs was 9-fold lower than that of free DOX solution in resistant P388/ADR cell line. In contrast, free IDA had comparable IC50 values as IDA NPs in Pgp-overexpressing P388/ADR and HCT-15 cells. In the in-vivo P388/ADR leukemia mouse model, the median survival time of DOX NPs was significantly greater than that of free DOX, and controls. In contrast, free IDA was equally as effective as IDA NPs in P388 and Pgp-overexpressing HCT-15 mouse tumor models. The cell uptake of IDA formulated as free IDA and IDA NPs was comparable in Pgp-overexpressing cells. In conclusion, DOX NPs could overcome Pgp-mediated MDR both in-vitro in P388/ADR leukemia cells and in-vivo in the murine leukemia mouse model. The present study suggests that our SLNs may offer potential to deliver anticancer drugs for the treatment of Pgp-mediated MDR in leukemia; however, selection of target drug may be very important.
这些研究的目的是调查和比较两种蒽环类抗生素,柔红霉素(IDA)和阿霉素(DOX)的固体脂质纳米粒(SLN),以对抗体外和体内使用不同的人和鼠癌细胞模型的 Pgp 介导的多药耐药(MDR)。IDA 和 DOX SLN 是从包含乳化蜡作为油相的温微乳液前体开发的,聚氧乙烯 20-硬脂醚(Brij 78)和 D-α-生育酚聚乙二醇琥珀酸酯(维生素 E TPGS)作为表面活性剂。阴离子离子对试剂,牛磺脱氧胆酸钠(STDC)和十四烷基硫酸钠(STS),用于中和阳离子蒽环类药物的电荷并增强药物在 SLN 中的包封。体外细胞毒性结果表明,DOX NPs 的 IC50 值比耐药 P388/ADR 细胞系中游离 DOX 溶液低 9 倍。相比之下,在 Pgp 过表达的 P388/ADR 和 HCT-15 细胞中,游离 IDA 与 IDA NPs 的 IC50 值相当。在体内 P388/ADR 白血病小鼠模型中,DOX NPs 的中位生存时间明显长于游离 DOX 和对照。相比之下,在 P388 和 Pgp 过表达的 HCT-15 小鼠肿瘤模型中,游离 IDA 与 IDA NPs 的疗效相当。在 Pgp 过表达细胞中,IDA 作为游离 IDA 和 IDA NPs 的细胞摄取相当。总之,DOX NPs 可以在体外 P388/ADR 白血病细胞中和体内白血病小鼠模型中克服 Pgp 介导的 MDR。本研究表明,我们的 SLN 可能为治疗白血病中的 Pgp 介导的 MDR 提供递送抗癌药物的潜力;然而,靶药的选择可能非常重要。