Tao Rong, Gong Jun, Luo Xixi, Zang Mengwei, Guo Wen, Wen Rong, Luo Zhijun
Department of Biochemistry, Boston University School of Medicine, 715 Albany Street, Evans 643, Boston, MA 02118, USA.
J Mol Signal. 2010 Feb 18;5(1):1. doi: 10.1186/1750-2187-5-1.
AMP-activated protein kinase (AMPK) is a fuel-sensing enzyme that is activated when cells experience energy deficiency and conversely suppressed in surfeit of energy supply. AMPK activation improves insulin sensitivity via multiple mechanisms, among which AMPK suppresses mTOR/S6K-mediated negative feedback regulation of insulin signaling.
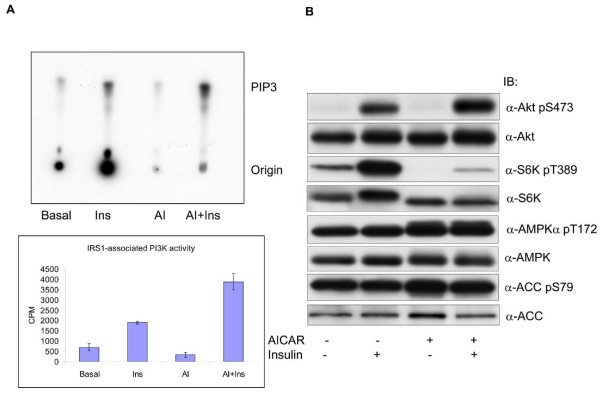
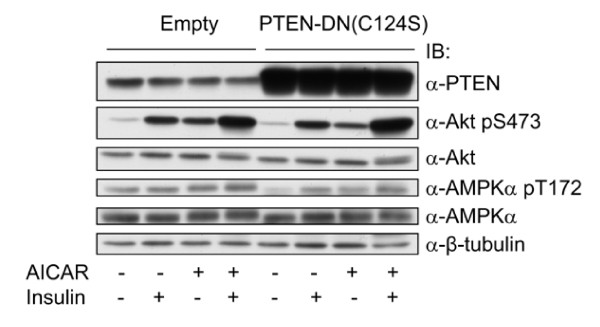
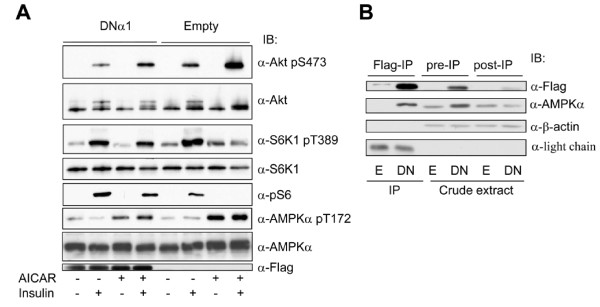
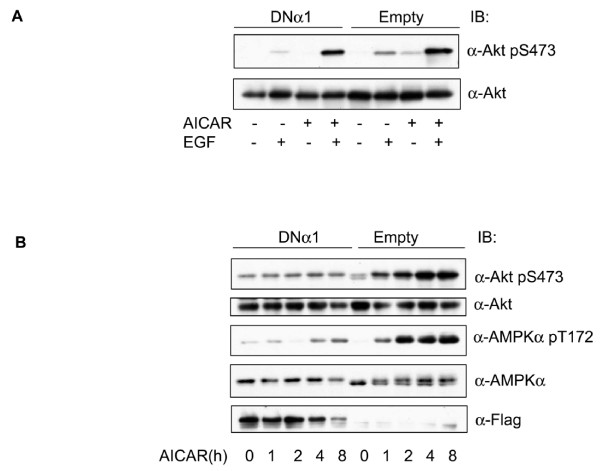
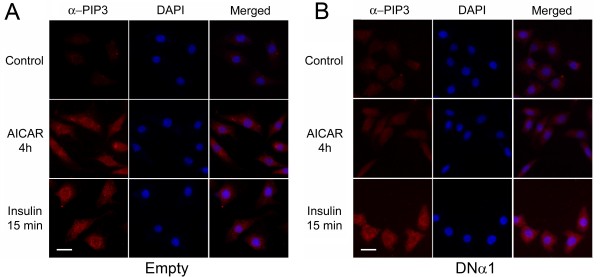
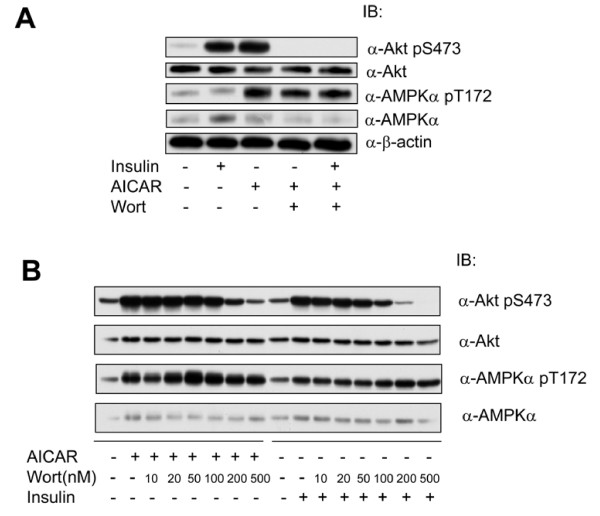
In the present study we further investigated the mechanism of AMPK-regulated insulin signaling. Our results showed that 5-aminoimidazole-4-carboxamide-1 ribonucleoside (AICAR) greatly enhanced the ability of insulin to stimulate the insulin receptor substrate-1 (IRS1)-associated PI3K activity in differentiated 3T3-F442a adipocytes, leading to increased Akt phosphorylation at S473, whereas insulin-stimulated activation of mTOR was diminished. In 3T3-F442a preadipocytes, these effects were attenuated by expression of a dominant negative mutant of AMPK alpha1 subunit. The enhancing effect of ACIAR on Akt phosphorylation was also observed when the cells were treated with EGF, suggesting that it is regulated at a step beyond IR/IRS1. Indeed, when the cells were chronically treated with AICAR in the absence of insulin, Akt phosphorylation was progressively increased. This event was associated with an increase in levels of phosphatidylinositol -3,4,5-trisphosphate (PIP3) and blocked by Wortmannin. We then expressed the dominant negative mutant of PTEN (C124S) and found that the inhibition of endogenous PTEN per se did not affect phosphorylation of Akt at basal levels or upon treatment with AICAR or insulin. Thus, this result suggests that AMPK activation of Akt is not mediated by regulating phosphatase and tensin homologue (PTEN).
Our present study demonstrates that AMPK exerts dual effects on the PI3K pathway, stimulating PI3K/Akt and inhibiting mTOR/S6K.
AMP激活的蛋白激酶(AMPK)是一种能量感应酶,当细胞能量不足时被激活,而在能量供应过剩时则相反被抑制。AMPK激活通过多种机制改善胰岛素敏感性,其中AMPK抑制mTOR/S6K介导的胰岛素信号负反馈调节。
在本研究中,我们进一步研究了AMPK调节胰岛素信号的机制。我们的结果表明,5-氨基咪唑-4-甲酰胺-1-核苷(AICAR)极大地增强了胰岛素刺激分化的3T3-F442a脂肪细胞中胰岛素受体底物-1(IRS1)相关PI3K活性的能力,导致Akt在S473处的磷酸化增加,而胰岛素刺激的mTOR激活则减弱。在3T3-F442a前脂肪细胞中,这些作用被AMPKα1亚基的显性负突变体的表达所减弱。当用表皮生长因子(EGF)处理细胞时,也观察到AICAR对Akt磷酸化的增强作用,这表明它在IR/IRS1之外的步骤受到调节。事实上,当细胞在没有胰岛素的情况下长期用AICAR处理时,Akt磷酸化逐渐增加。这一事件与磷脂酰肌醇-3,4,5-三磷酸(PIP3)水平的增加有关,并被渥曼青霉素所阻断。然后我们表达了PTEN(C124S)的显性负突变体,发现抑制内源性PTEN本身并不影响基础水平或用AICAR或胰岛素处理时Akt的磷酸化。因此,这一结果表明AMPK对Akt的激活不是通过调节磷酸酶和张力蛋白同源物(PTEN)介导的。
我们目前的研究表明,AMPK对PI3K途径发挥双重作用,刺激PI3K/Akt并抑制mTOR/S6K。