Wang Yingli, Sun Miao, Uhlhorn Victoria L, Zhou Xueyan, Peter Inga, Martinez-Abadias Neus, Hill Cheryl A, Percival Christopher J, Richtsmeier Joan T, Huso David L, Jabs Ethylin Wang
Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, New York, USA.
BMC Dev Biol. 2010 Feb 22;10:22. doi: 10.1186/1471-213X-10-22.
Apert syndrome is characterized by craniosynostosis and limb abnormalities and is primarily caused by FGFR2 +/P253R and +/S252W mutations. The former mutation is present in approximately one third whereas the latter mutation is present in two-thirds of the patients with this condition. We previously reported an inbred transgenic mouse model with the Fgfr2 +/S252W mutation on the C57BL/6J background for Apert syndrome. Here we present a mouse model for the Fgfr2+/P253R mutation.
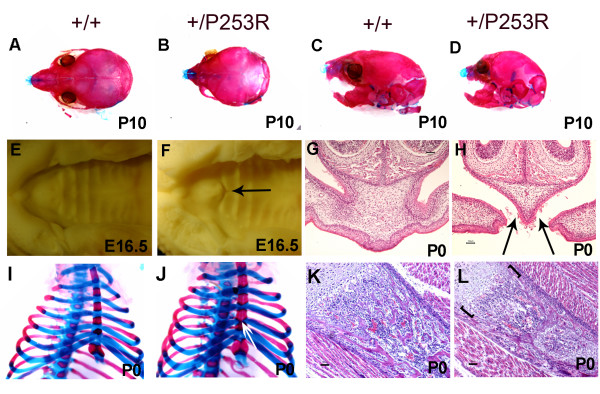
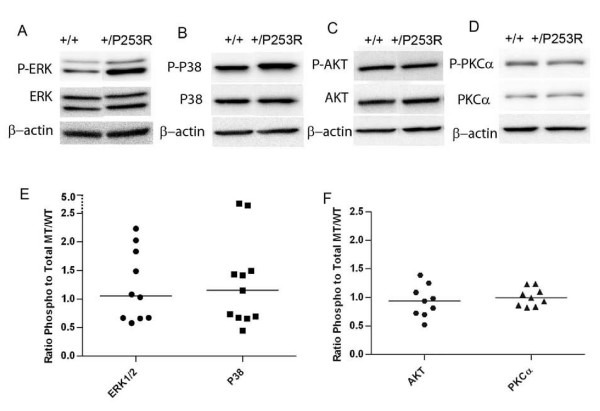
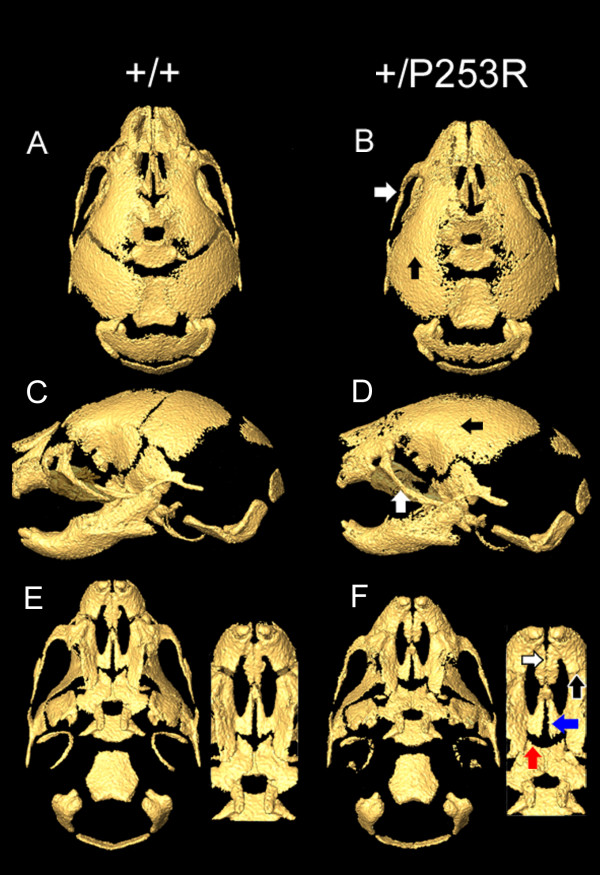
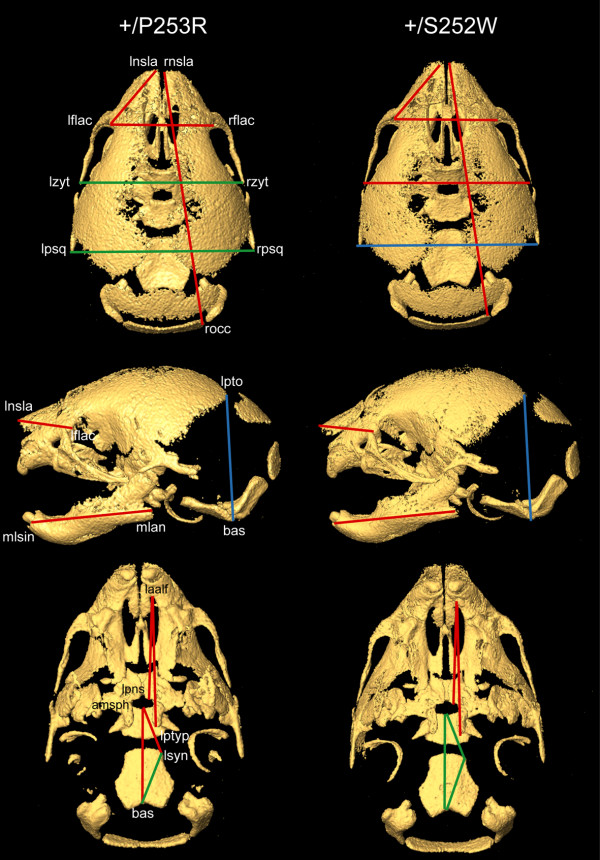
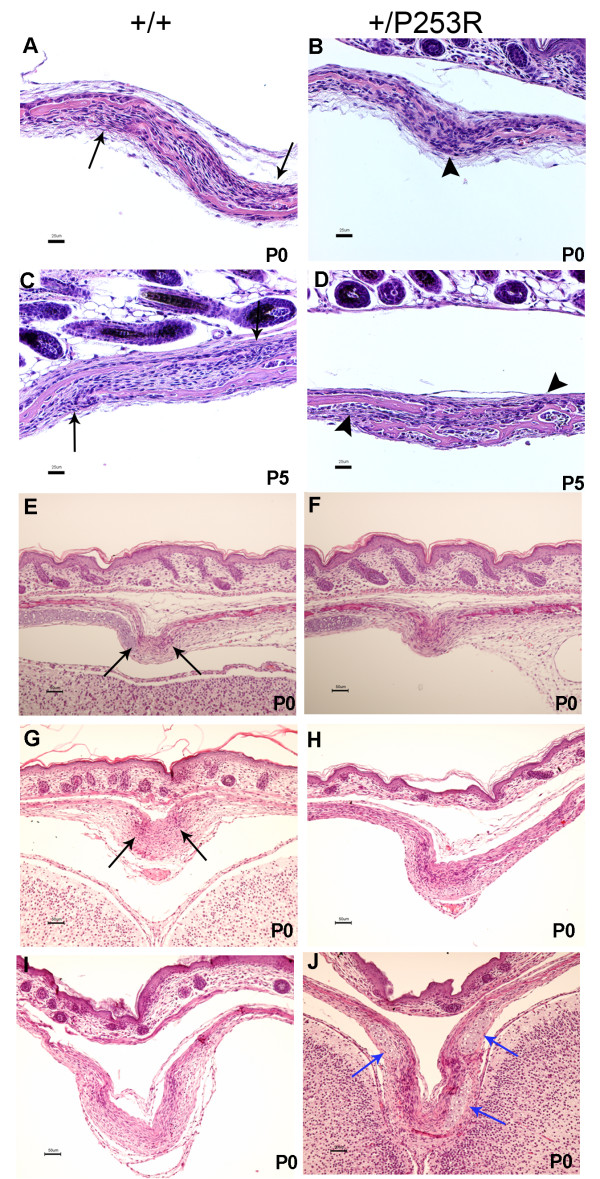
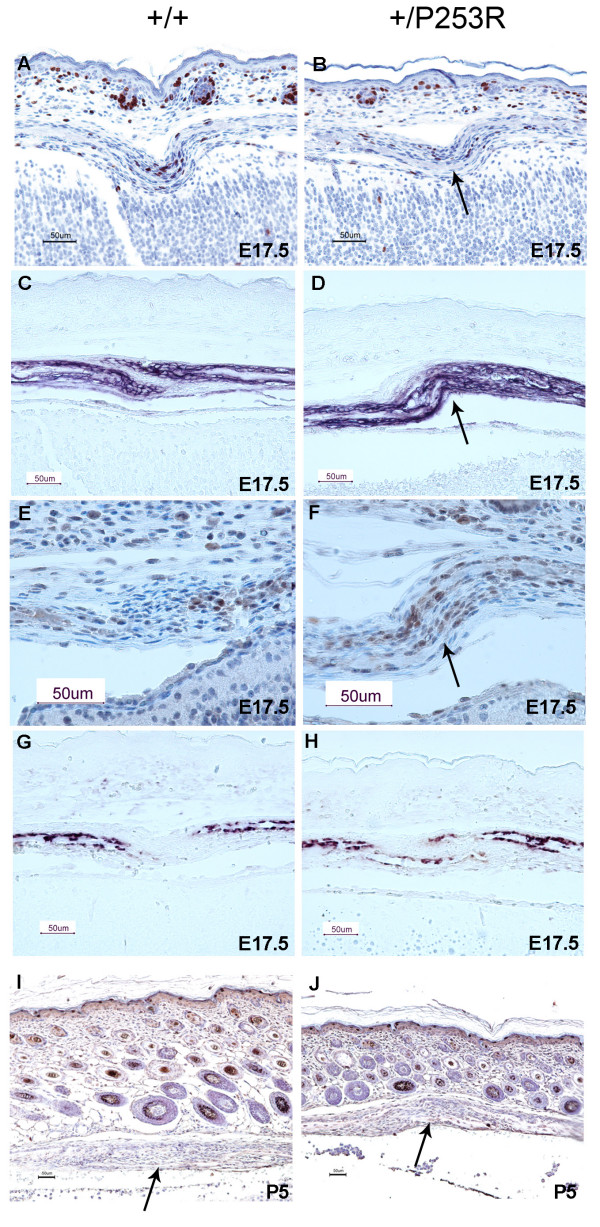
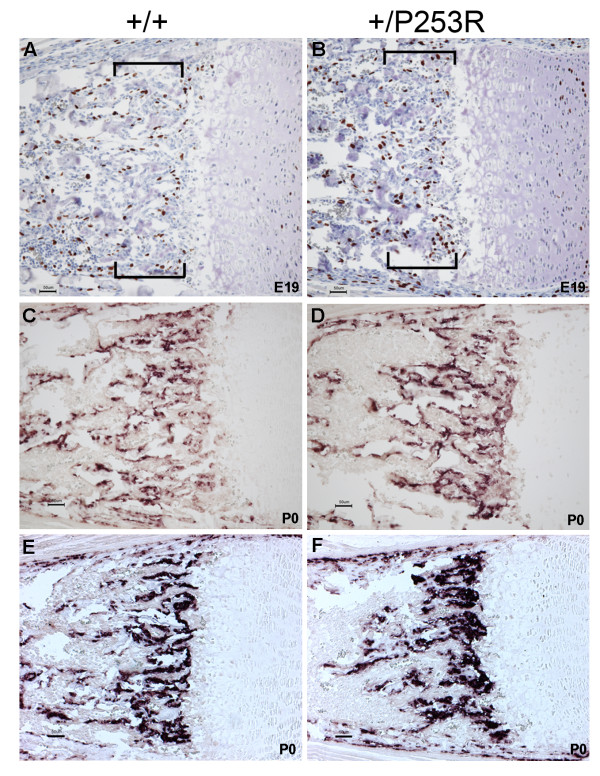
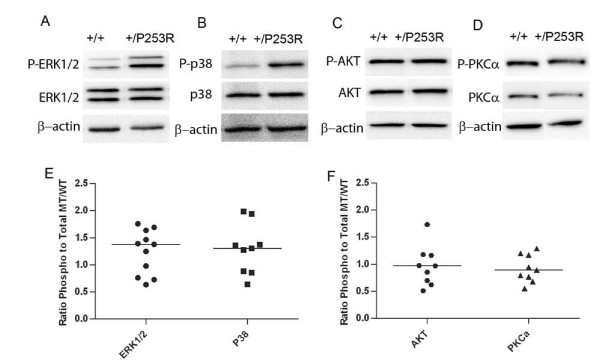
We generated inbred Fgfr2(+/P253R) mice on the same C56BL/6J genetic background and analyzed their skeletal abnormalities. 3D micro-CT scans of the skulls of the Fgfr2(+/P253R) mice revealed that the skull length was shortened with the length of the anterior cranial base significantly shorter than that of the Fgfr2(+/S252W) mice at P0. The Fgfr2(+/P253R) mice presented with synostosis of the coronal suture and proximate fronts with disorganized cellularity in sagittal and lambdoid sutures. Abnormal osteogenesis and proliferation were observed at the developing coronal suture and long bones of the Fgfr2(+/P253R) mice as in the Fgfr2(+/S252W) mice. Activation of mitogen-activated protein kinases (MAPK) was observed in the Fgfr2(+/P253R) neurocranium with an increase in phosphorylated p38 as well as ERK1/2, whereas phosphorylated AKT and PKCalpha were not obviously changed as compared to those of wild-type controls. There were localized phenotypic and molecular variations among individual embryos with different mutations and among those with the same mutation.
Our in vivo studies demonstrated that the Fgfr2 +/P253R mutation resulted in mice with cranial features that resemble those of the Fgfr2(+/S252W) mice and human Apert syndrome. Activated p38 in addition to the ERK1/2 signaling pathways may mediate the mutant neurocranial phenotype. Though Apert syndrome is traditionally thought to be a consistent phenotype, our results suggest localized and regional variations in the phenotypes that characterize Apert syndrome.
Apert综合征的特征为颅缝早闭和肢体异常,主要由FGFR2 +/P253R和+/S252W突变引起。前一种突变存在于约三分之一的患者中,而后一种突变存在于三分之二的该疾病患者中。我们之前报道了一种在C57BL/6J背景上携带Fgfr2 +/S252W突变的近交转基因小鼠模型用于研究Apert综合征。在此,我们展示一种针对Fgfr2+/P253R突变的小鼠模型。
我们在相同的C56BL/6J遗传背景上培育出近交Fgfr2(+/P253R)小鼠,并分析它们的骨骼异常情况。对Fgfr2(+/P253R)小鼠颅骨进行的三维显微CT扫描显示,在出生后第0天,颅骨长度缩短,前颅底长度显著短于Fgfr2(+/S252W)小鼠。Fgfr2(+/P253R)小鼠出现冠状缝和邻近额缝的骨缝早闭,矢状缝和人字缝细胞排列紊乱。在Fgfr2(+/P253R)小鼠发育中的冠状缝和长骨中观察到与Fgfr2(+/S^252W)小鼠一样的异常成骨和增殖。在Fgfr2(+/P253R)神经颅骨中观察到丝裂原活化蛋白激酶(MAPK)的激活,磷酸化的p38以及ERK1/2增加,而与野生型对照相比,磷酸化的AKT和PKCalpha没有明显变化。在具有不同突变的个体胚胎之间以及具有相同突变的胚胎之间存在局部表型和分子差异。
我们的体内研究表明,Fgfr'2 +/P253R突变导致小鼠出现类似于Fgfr2(+/S252W)小鼠和人类Apert综合征的颅骨特征。除ERK1/2信号通路外,激活的p38可能介导突变的神经颅骨表型。尽管传统上认为Apert综合征是一种一致的表型,但我们的结果表明Apert综合征特征性表型存在局部和区域差异。