Department of Pharmacology, University of Melbourne, Victoria, Australia.
Respir Res. 2010 Feb 23;11(1):21. doi: 10.1186/1465-9921-11-21.
Transforming growth factor beta1 (TGF-beta1)-mediated epithelial mesenchymal transition (EMT) of alveolar epithelial cells (AEC) may contribute to lung fibrosis. Since PPAR gamma ligands have been shown to inhibit fibroblast activation by TGF-beta1, we assessed the ability of the thiazolidinediones rosiglitazone (RGZ) and ciglitazone (CGZ) to regulate TGF-beta1-mediated EMT of A549 cells, assessing changes in cell morphology, and expression of cell adhesion molecules E-cadherin (epithelial cell marker) and N-cadherin (mesenchymal cell marker), and collagen 1 alpha 1 (COL1A1), CTGF and MMP-2 mRNA.
Serum-deprived A549 cells (human AEC cell line) were pre-incubated with RGZ and CGZ (1 - 30 microM) in the absence or presence of the PPAR gamma antagonist GW9662 (10 microM) before TGFbeta-1 (0.075-7.5 ng/ml) treatment for up to 72 hrs. Changes in E-cadherin, N-cadherin and phosphorylated Smad2 and Smad3 levels were analysed by Western blot, and changes in mRNA levels including COL1A1 assessed by RT-PCR.
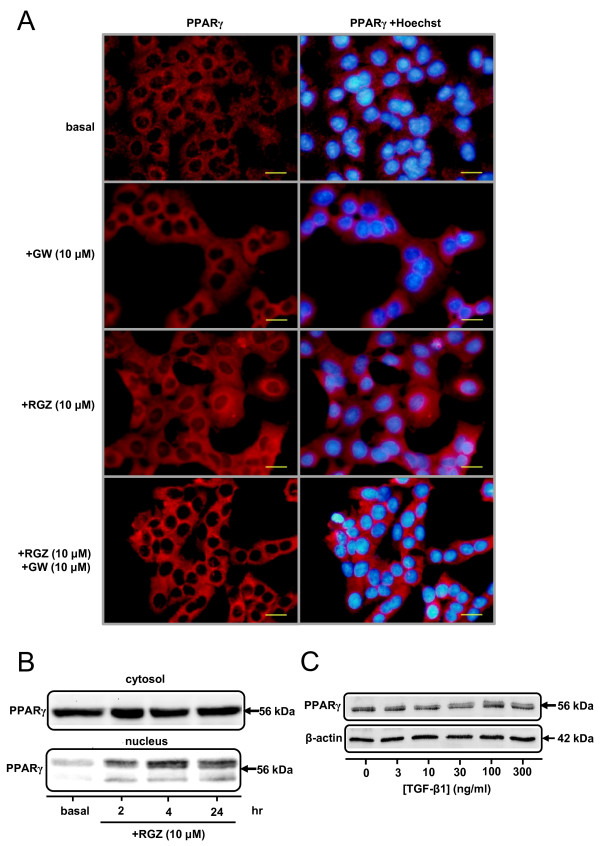
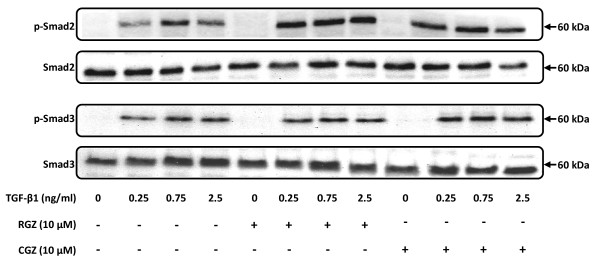
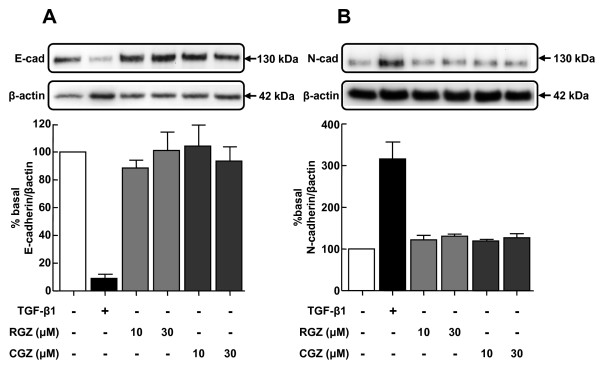
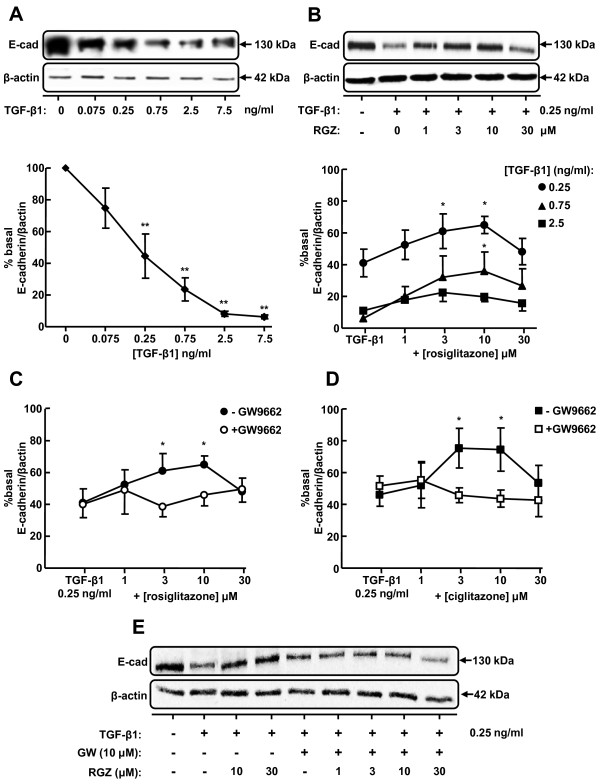
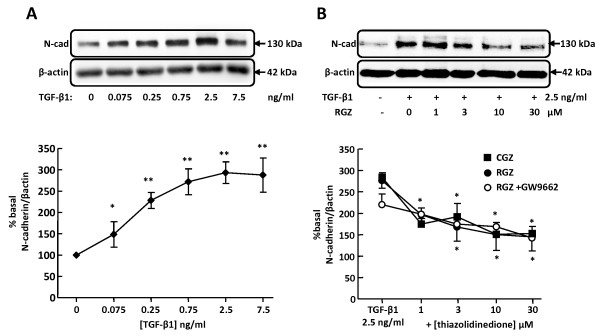
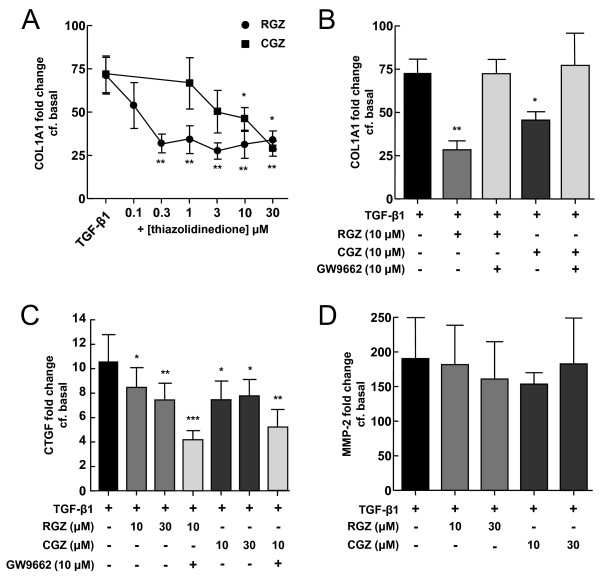
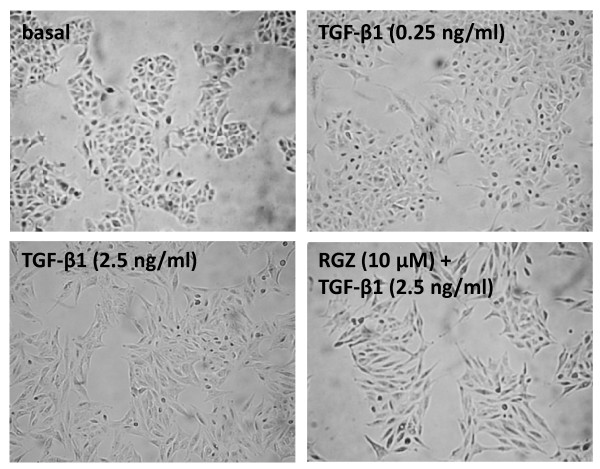
TGFbeta-1 (2.5 ng/ml)-induced reductions in E-cadherin expression were associated with a loss of epithelial morphology and cell-cell contact. Concomitant increases in N-cadherin, MMP-2, CTGF and COL1A1 were evident in predominantly elongated fibroblast-like cells. Neither RGZ nor CGZ prevented TGF beta 1-induced changes in cell morphology, and PPAR gamma-dependent inhibitory effects of both ligands on changes in E-cadherin were only evident at submaximal TGF-beta1 (0.25 ng/ml). However, both RGZ and CGZ inhibited the marked elevation of N-cadherin and COL1A1 induced by TGF-beta1 (2.5 ng/ml), with effects on COL1A1 prevented by GW9662. Phosphorylation of Smad2 and Smad3 by TGF-beta1 was not inhibited by RGZ or CGZ.
RGZ and CGZ inhibited profibrotic changes in TGF-beta1-stimulated A549 cells independently of inhibition of Smad phosphorylation. Their inhibitory effects on changes in collagen I and E-cadherin, but not N-cadherin or CTGF, appeared to be PPAR gamma-dependent. Further studies are required to unravel additional mechanisms of inhibition of TGF-beta1 signalling by thiazolidinediones and their implications for the contribution of EMT to lung fibrosis.
转化生长因子-β 1(TGF-β1)介导的肺泡上皮细胞(AEC)上皮间质转化(EMT)可能导致肺纤维化。由于过氧化物酶体增殖物激活受体γ配体已被证明可抑制 TGF-β1 激活的成纤维细胞,我们评估了噻唑烷二酮类药物罗格列酮(RGZ)和吡格列酮(CGZ)调节 A549 细胞 TGF-β1 介导的 EMT 的能力,评估了细胞形态变化以及细胞黏附分子 E-钙粘蛋白(上皮细胞标志物)和 N-钙粘蛋白(间充质细胞标志物)和胶原 1α1(COL1A1)、CTGF 和 MMP-2mRNA 的表达。
在无血清培养的 A549 细胞(人 AEC 细胞系)中,在 TGFβ-1(0.075-7.5ng/ml)处理之前,用 RGZ 和 CGZ(1-30μM)在不存在或存在过氧化物酶体增殖物激活受体γ拮抗剂 GW9662(10μM)的情况下预先孵育,长达 72 小时。通过 Western blot 分析 E-钙粘蛋白、N-钙粘蛋白和磷酸化 Smad2 和 Smad3 水平的变化,并通过 RT-PCR 评估 COL1A1 等 mRNA 水平的变化。
TGF-β1(2.5ng/ml)诱导的 E-钙粘蛋白表达减少与上皮形态和细胞-细胞接触的丧失有关。在主要呈长形成纤维样细胞中,同时观察到 N-钙粘蛋白、MMP-2、CTGF 和 COL1A1 的增加。RGZ 和 CGZ 均不能防止 TGF-β1 诱导的细胞形态变化,两种配体对 E-钙粘蛋白变化的 PPARγ依赖性抑制作用仅在亚最大 TGF-β1(0.25ng/ml)时才明显。然而,RGZ 和 CGZ 均抑制了 TGF-β1(2.5ng/ml)诱导的 N-钙粘蛋白和 COL1A1 的明显升高,GW9662 可预防 COL1A1 的升高。RGZ 和 CGZ 均未抑制 TGF-β1 诱导的 Smad2 和 Smad3 的磷酸化。
RGZ 和 CGZ 抑制 TGF-β1 刺激的 A549 细胞的促纤维化变化,而不抑制 Smad 磷酸化。它们对胶原 I 和 E-钙粘蛋白变化的抑制作用,但不是 N-钙粘蛋白或 CTGF,似乎依赖于过氧化物酶体增殖物激活受体γ。需要进一步的研究来揭示噻唑烷二酮类药物抑制 TGF-β1 信号的其他机制及其对 EMT 对肺纤维化的贡献的影响。