Department of Biochemistry and Molecular Biology, University of Southern California, Los Angeles, California, United States of America.
PLoS One. 2010 Feb 22;5(2):e9359. doi: 10.1371/journal.pone.0009359.
Epithelial ovarian carcinoma is a significant cause of cancer mortality in women worldwide and in the United States. Epithelial ovarian cancer comprises several histological subtypes, each with distinct clinical and molecular characteristics. The natural history of this heterogeneous disease, including the cell types of origin, is poorly understood. This study applied recently developed methods for high-throughput DNA methylation profiling to characterize ovarian cancer cell lines and tumors, including representatives of three major histologies.
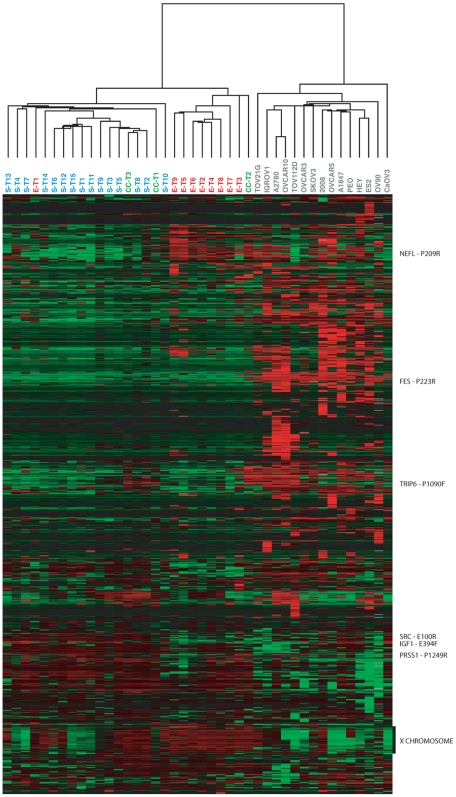
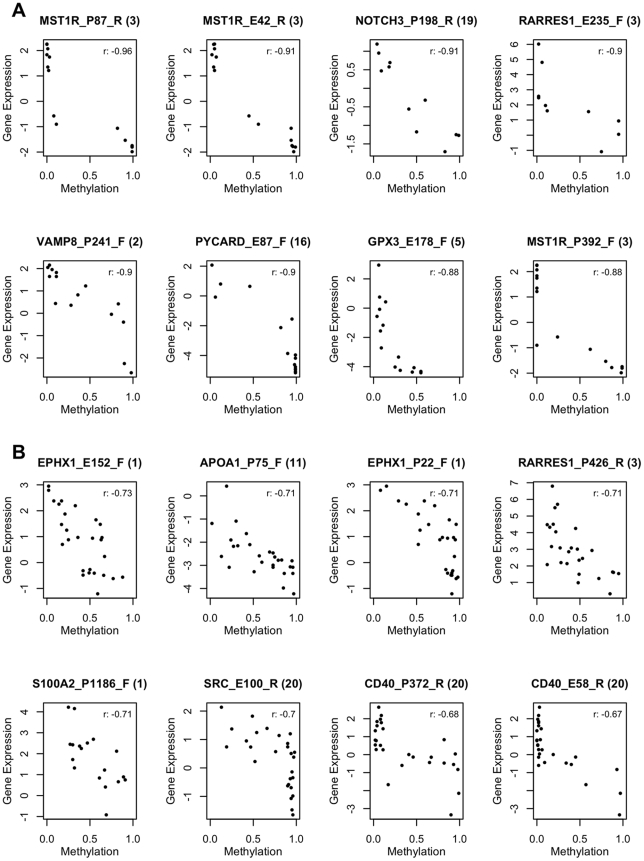
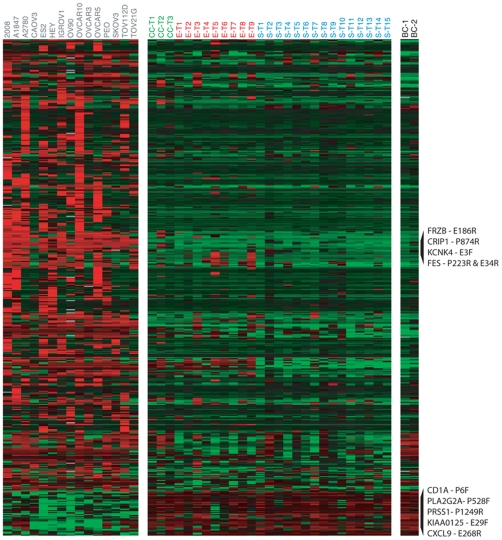
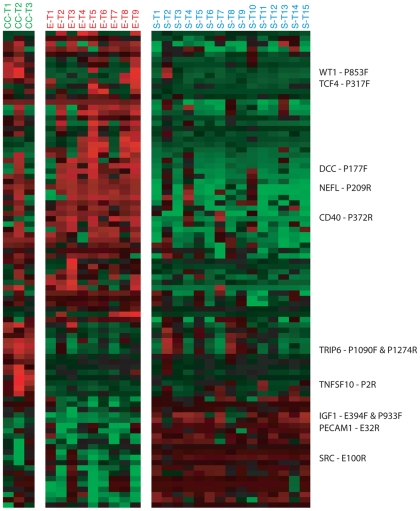
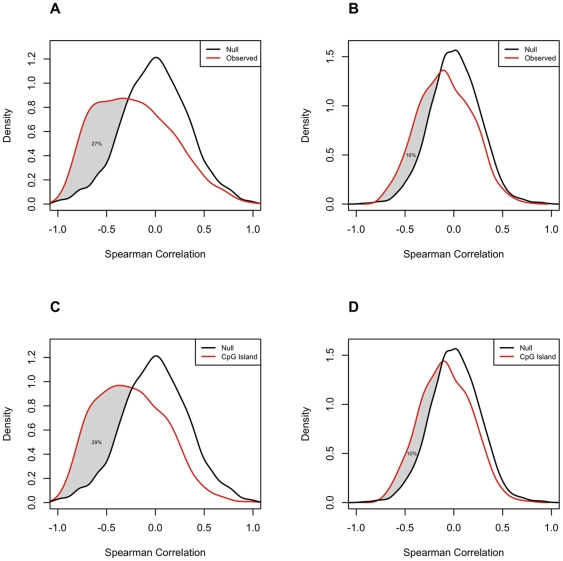
METHODOLOGY/PRINCIPAL FINDINGS: We obtained DNA methylation profiles of 1,505 CpG sites (808 genes) in 27 primary epithelial ovarian tumors and 15 ovarian cancer cell lines. We found that the DNA methylation profiles of ovarian cancer cell lines were markedly different from those of primary ovarian tumors. Aggregate DNA methylation levels of the assayed CpG sites tended to be higher in ovarian cancer cell lines relative to ovarian tumors. Within the primary tumors, those of the same histological type were more alike in their methylation profiles than those of different subtypes. Supervised analyses identified 90 CpG sites (68 genes) that exhibited 'subtype-specific' DNA methylation patterns (FDR<1%) among the tumors. In ovarian cancer cell lines, we estimated that for at least 27% of analyzed autosomal CpG sites, increases in methylation were accompanied by decreases in transcription of the associated gene.
The significant difference in DNA methylation profiles between ovarian cancer cell lines and tumors underscores the need to be cautious in using cell lines as tumor models for molecular studies of ovarian cancer and other cancers. Similarly, the distinct methylation profiles of the different histological types of ovarian tumors reinforces the need to treat the different histologies of ovarian cancer as different diseases, both clinically and in biomarker studies. These data provide a useful resource for future studies, including those of potential tumor progenitor cells, which may help illuminate the etiology and natural history of these cancers.
上皮性卵巢癌是全球和美国女性癌症死亡的重要原因。上皮性卵巢癌包括几种组织学亚型,每种亚型都具有独特的临床和分子特征。这种异质性疾病的自然史,包括起源细胞类型,了解甚少。本研究应用最近开发的高通量 DNA 甲基化分析方法,对包括三种主要组织学类型代表在内的卵巢癌细胞系和肿瘤进行了特征描述。
方法/主要发现:我们获得了 27 例原发性上皮性卵巢肿瘤和 15 例卵巢癌细胞系中 1505 个 CpG 位点(808 个基因)的 DNA 甲基化谱。我们发现卵巢癌细胞系的 DNA 甲基化谱与原发性卵巢肿瘤明显不同。所检测 CpG 位点的总 DNA 甲基化水平在卵巢癌细胞系中相对卵巢肿瘤更高。在原发性肿瘤中,同一组织学类型的肿瘤在其甲基化谱中比不同亚型的肿瘤更为相似。有监督分析确定了 90 个 CpG 位点(68 个基因),它们在肿瘤中表现出“亚型特异性”的 DNA 甲基化模式(FDR<1%)。在卵巢癌细胞系中,我们估计至少 27%的分析常染色体 CpG 位点,其甲基化增加伴随着相关基因转录的减少。
卵巢癌细胞系和肿瘤之间 DNA 甲基化谱的显著差异突出表明,在进行卵巢癌和其他癌症的分子研究时,必须谨慎使用细胞系作为肿瘤模型。同样,不同组织学类型的卵巢肿瘤的不同甲基化谱也强调了需要将卵巢癌的不同组织学类型视为不同的疾病,无论是在临床还是在生物标志物研究中。这些数据为未来的研究提供了有用的资源,包括对潜在肿瘤祖细胞的研究,这可能有助于阐明这些癌症的病因和自然史。