Nof Eyal, Cordeiro Jonathan M, Pérez Guillermo J, Scornik Fabiana S, Calloe Kirstine, Love Barry, Burashnikov Elena, Caceres Gabriel, Gunsburg Moshe, Antzelevitch Charles
Masonic Medical Research Laboratory, Utica, NY 13501, USA.
Circ Cardiovasc Genet. 2010 Apr;3(2):199-206. doi: 10.1161/CIRCGENETICS.109.898569. Epub 2010 Feb 24.
Identification of infants at risk for sudden arrhythmic death remains one of the leading challenges of modern medicine. We present a family in which a common polymorphism (single nucleotide polymorphism) inherited from the father, combined with a stop codon mutation inherited from the mother (both asymptomatic), led to 2 cases of sudden infant death.
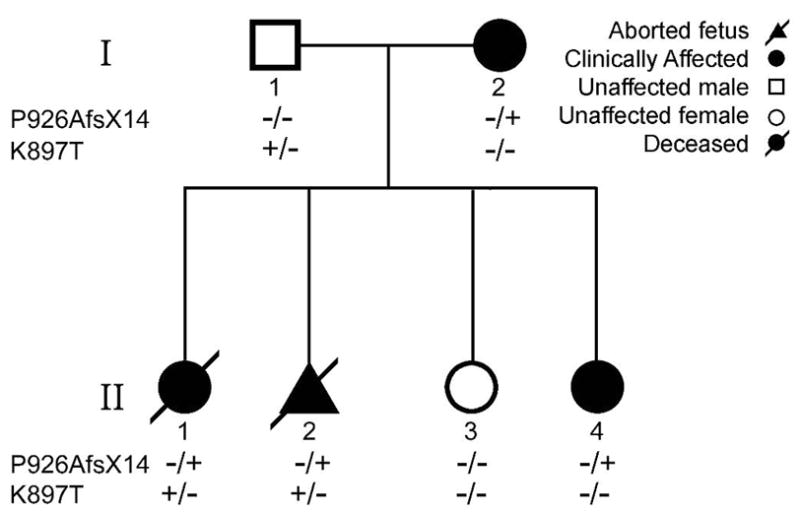
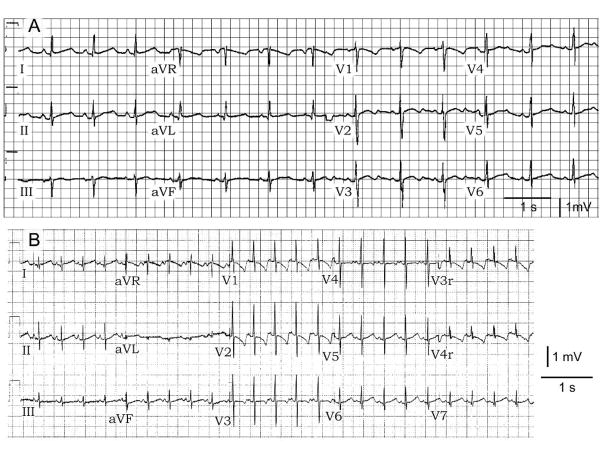
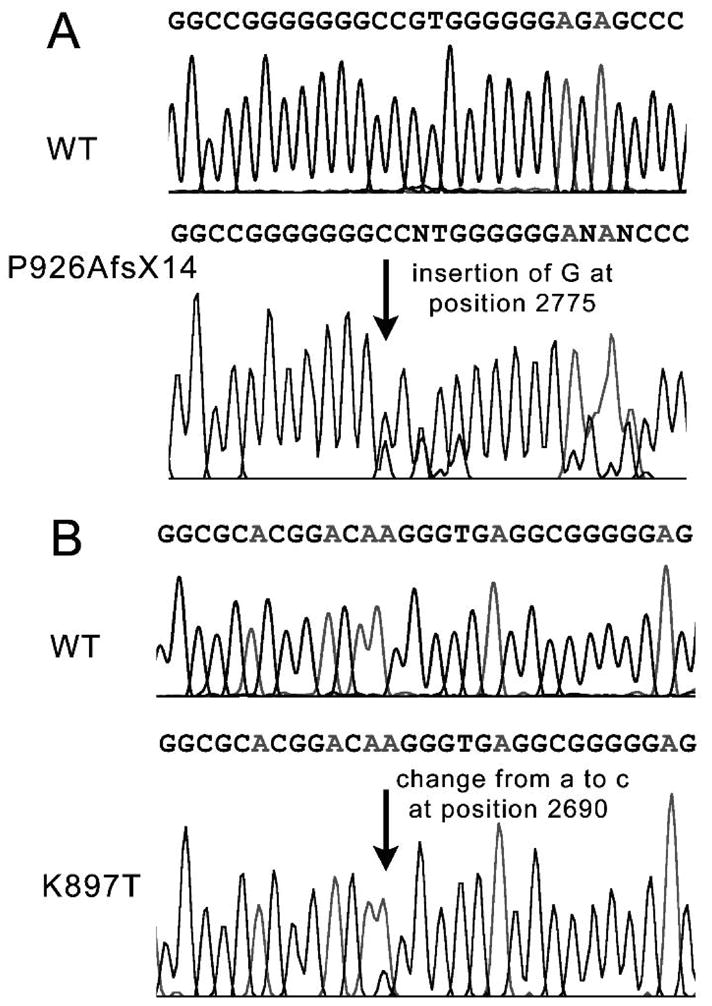
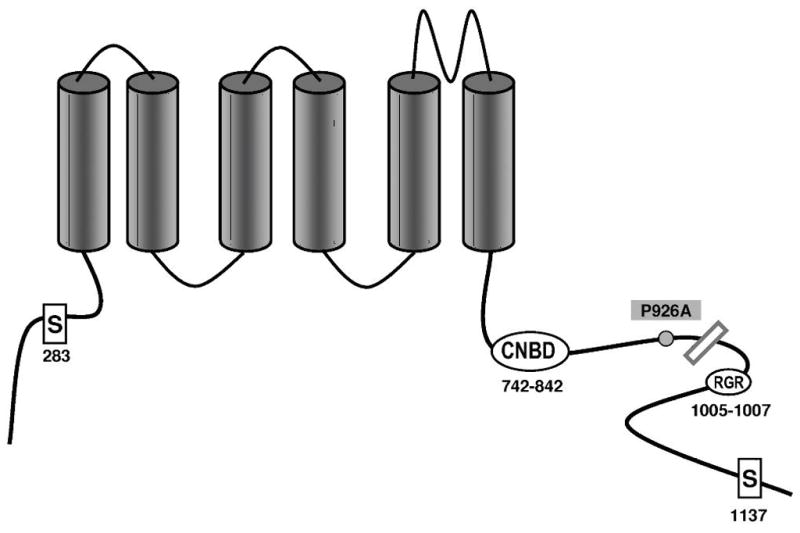
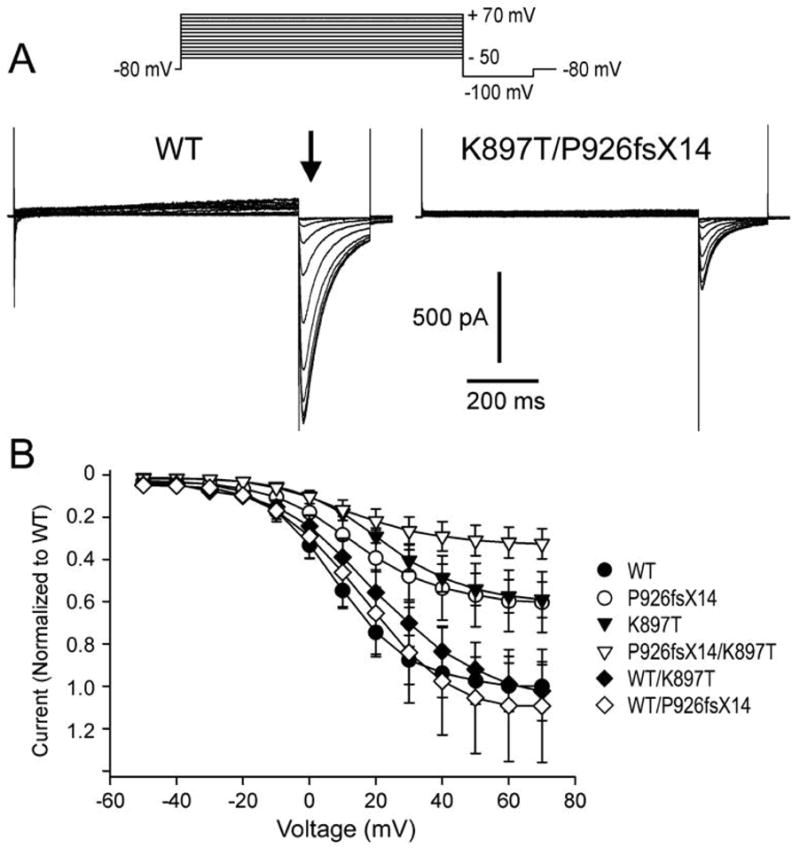
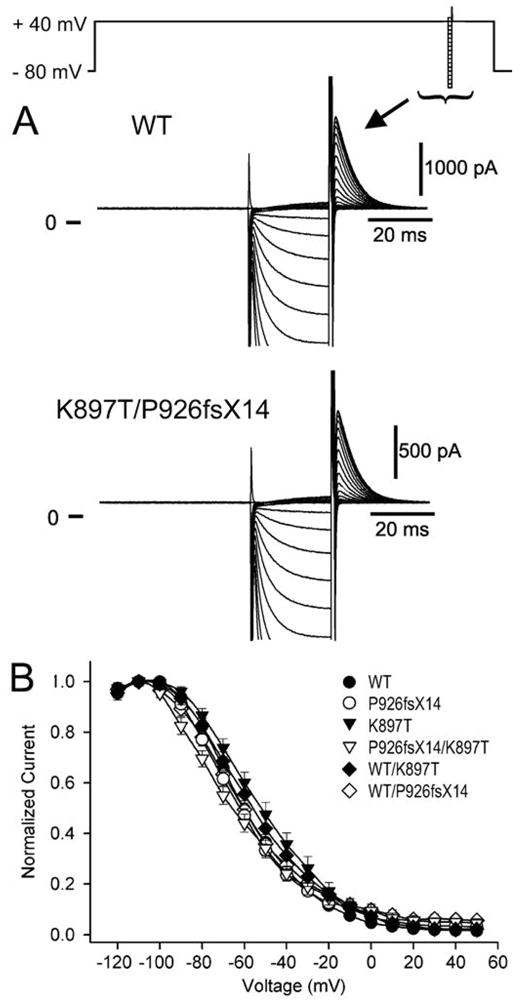
KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, CACNA1c, CACNB2b, and KCNJ2 genes were amplified and analyzed by direct sequencing. Functional electrophysiological studies were performed with the single nucleotide polymorphism and mutation expressed singly and in combination in Chinese ovary (CHO-K1) and COS-1 cells. An asymptomatic woman presenting after the death of her 2-day-old infant and spontaneous abortion of a second baby in the first trimester was referred for genetic analysis. The newborn infant had nearly incessant ventricular tachycardia while in utero and a prolonged QTc (560 ms). The mother was asymptomatic but displayed a prolonged QTc. Genetic screening of the mother revealed a heterozygous nonsense mutation (P926AfsX14) in KCNH2, predicting a stop codon. The father was asymptomatic with a normal QTc but had a heterozygous polymorphism (K897T) in KCNH2. The baby who died at 2 days of age and the aborted fetus inherited both K897T and P926AfsX14. Heterologous coexpression of K897T and P926AfsX14 led to loss of function of HERG current much greater than expression of K897T or P926AfsX14 alone.
Our data suggest that a common polymorphism (K897T) can markedly accentuate the loss of function of mildly defective HERG channels, leading to long-QT syndrome-mediated arrhythmias and sudden infant death.
识别有猝死风险的婴儿仍然是现代医学面临的主要挑战之一。我们报道了一个家庭,父亲遗传的常见多态性(单核苷酸多态性)与母亲遗传的终止密码子突变(二者均无症状)相结合,导致了两例婴儿猝死。
对KCNQ1、KCNH2、SCN5A、KCNE1、KCNE2、CACNA1c、CACNB2b和KCNJ2基因进行扩增并通过直接测序进行分析。在中国仓鼠卵巢(CHO-K1)细胞和COS-1细胞中单独及联合表达单核苷酸多态性和突变,进行功能性电生理研究。一名2日龄婴儿死亡且孕早期第二个胎儿自然流产后前来就诊的无症状女性接受了基因分析。该新生儿在子宫内时几乎持续存在室性心动过速,QTc延长(560毫秒)。母亲无症状,但QTc延长。对母亲的基因筛查显示KCNH2存在杂合性无义突变(P926AfsX14),预测有一个终止密码子。父亲无症状,QTc正常,但KCNH2存在杂合性多态性(K897T)。2日龄死亡的婴儿和流产胎儿均遗传了K897T和P926AfsX14。K897T和P926AfsX14的异源共表达导致HERG电流功能丧失的程度远大于单独表达K897T或P926AfsX14。
我们的数据表明,常见的多态性(K897T)可显著加重轻度缺陷的HERG通道的功能丧失程度,导致长QT综合征介导的心性心律失常和婴儿猝死。