Chiabrando Deborah, Tolosano Emanuela
Molecular Biotechnology Center, University of Torino, Via Nizza 52, 10126 Torino, Italy.
Adv Hematol. 2010;2010:790632. doi: 10.1155/2010/790632. Epub 2010 May 5.
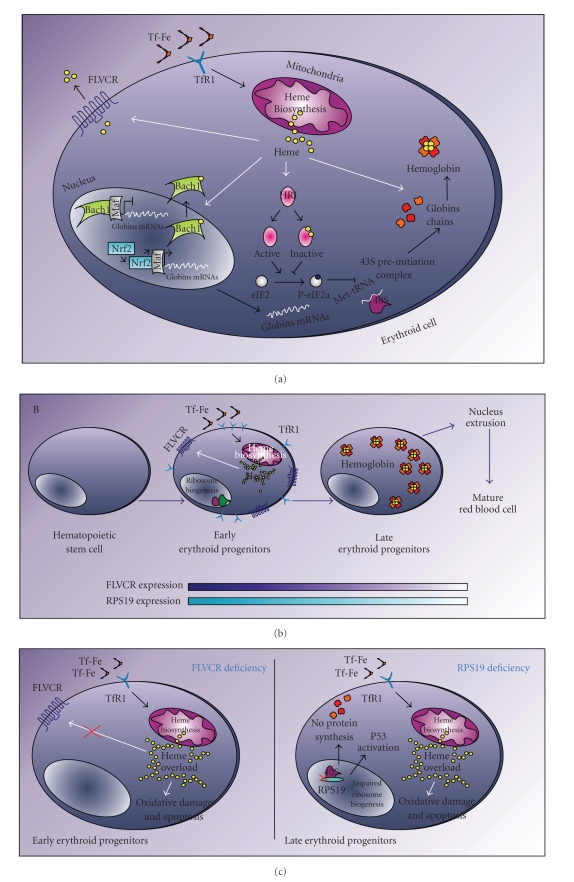
Diamond-Blackfan anemia (DBA) is a rare, pure red-cell aplasia that presents during infancy. Approximately 40% of cases are associated with other congenital defects, particularly malformations of the upper limb or craniofacial region. Mutations in the gene coding for the ribosomal protein RPS19 have been identified in 25% of patients with DBA, with resulting impairment of 18S rRNA processing and 40S ribosomal subunit formation. Moreover, mutations in other ribosomal protein coding genes account for about 25% of other DBA cases. Recently, the analysis of mice from which the gene coding for the heme exporter Feline Leukemia Virus subgroup C Receptor (FLVCR1) is deleted suggested that this gene may be involved in the pathogenesis of DBA. FLVCR1-null mice show a phenotype resembling that of DBA patients, including erythroid failure and malformations. Interestingly, some DBA patients have disease linkage to chromosome 1q31, where FLVCR1 is mapped. Moreover, it has been reported that cells from DBA patients express alternatively spliced isoforms of FLVCR1 which encode non-functional proteins. Herein, we review the known roles of RPS19 and FLVCR1 in ribosome function and heme metabolism respectively, and discuss how the deficiency of a ribosomal protein or of a heme exporter may result in the same phenotype.
先天性纯红细胞再生障碍性贫血(DBA)是一种罕见的、在婴儿期出现的纯红细胞再生障碍。约40%的病例与其他先天性缺陷有关,特别是上肢或颅面部畸形。在25%的DBA患者中已发现编码核糖体蛋白RPS19的基因突变,导致18S rRNA加工和40S核糖体亚基形成受损。此外,其他核糖体蛋白编码基因的突变约占其他DBA病例的25%。最近,对编码血红素输出蛋白猫白血病病毒C亚群受体(FLVCR1)的基因被删除的小鼠的分析表明,该基因可能参与DBA的发病机制。FLVCR1基因敲除小鼠表现出类似于DBA患者的表型,包括红细胞生成障碍和畸形。有趣的是,一些DBA患者的疾病与FLVCR1所在的1q31染色体存在连锁关系。此外,据报道,DBA患者的细胞表达FLVCR1的可变剪接异构体,其编码无功能的蛋白质。在此,我们分别综述了RPS19和FLVCR1在核糖体功能和血红素代谢中的已知作用,并讨论了核糖体蛋白或血红素输出蛋白的缺乏如何导致相同的表型。