School of Post-baccalaureate Chinese Medicine, China Medical University, Hsueh-Shih Rd,, Taichung, Taiwan.
J Biomed Sci. 2010 Sep 30;17(1):79. doi: 10.1186/1423-0127-17-79.
GM1 gangliosidosis (GM1) is an autosomal recessive lysosomal storage disease caused by deficiency of acid beta-galactosidase (GLB1; EC3.2.1.23). Here, we identify three novel mutations in the GLB1 gene from two Han Chinese patients with GM1 that appear correlated with clinical phenotype.
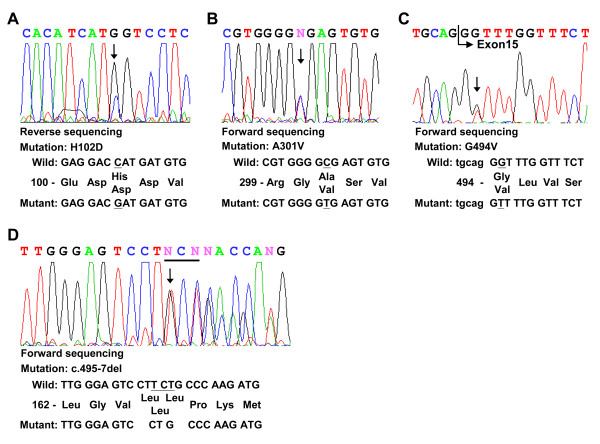
One of the two Han Chinese patients with GM1 presented with the juvenile form, and the other with the infantile form with cardiac involvement. Sequencing of the entire GLB1 gene revealed three novel mutations (p.H102 D, p.G494V, c.495_497delTCT), which were absent in 94 normal controls. Transient expression of cDNA encoding these variants was performed in COS-1 cells to evaluate β-galactosidase activities.
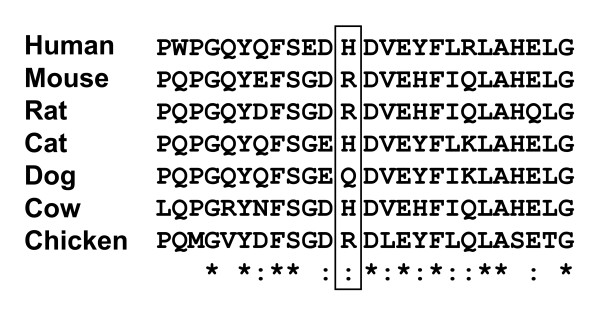
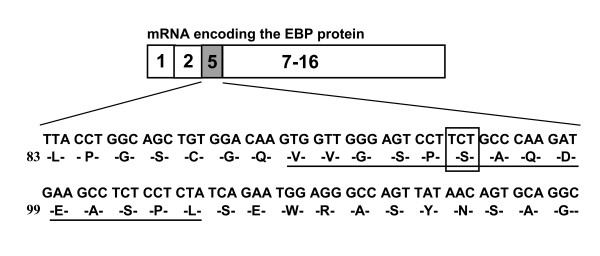
The first case (patient 1) with the juvenile form contained two missense mutations, p.H102 D and p.A301V. Patient 2 diagnosed with the infantile form of the disease with cardiac involvement was compound heterozygous for p.G494V and c.495_497delTCT mutations. All mutant beta-galactosidases exhibited significantly reduced activity (12%, 0%, 0%, and 0% for p.H102 D, p.A301V, p.G494V, and c.495_497delTCT), compared with the wild-type beta-galactosidase cDNA clone. The mutations identified in patient 2 with cardiomyopathy were localized in the GLB1 gene region common to both lysosomal beta-galactosidase and elastin binding protein (EBP), and caused a deletion in the elastin-binding domain of EBP.
All four mutations identified in Han Chinese patients induce significant suppression of β-galactosidase activity, correlating with severity of disease and presence of cardiomyopathy.
GM1 神经节苷脂贮积症(GM1)是一种常染色体隐性溶酶体贮积病,由酸性β-半乳糖苷酶(GLB1;EC3.2.1.23)缺乏引起。在这里,我们从两名 GM1 汉族患者中鉴定出 GLB1 基因中的三个新突变,这些突变似乎与临床表型相关。
两名 GM1 汉族患者之一表现为少年型,另一名表现为伴有心脏受累的婴儿型。对整个 GLB1 基因进行测序,发现了三个新突变(p.H102 D、p.G494V、c.495_497delTCT),这些突变在 94 名正常对照中均不存在。在 COS-1 细胞中瞬时表达编码这些变体的 cDNA,以评估β-半乳糖苷酶活性。
首例(患者 1)为少年型,包含两个错义突变,p.H102 D 和 p.A301V。诊断为伴有心脏受累的婴儿型疾病的患者 2 为 p.G494V 和 c.495_497delTCT 突变的复合杂合子。与野生型β-半乳糖苷酶 cDNA 克隆相比,所有突变型β-半乳糖苷酶的活性均显著降低(p.H102 D、p.A301V、p.G494V 和 c.495_497delTCT 的活性分别为 12%、0%、0%和 0%)。在伴有心肌病的患者 2 中发现的突变定位于 GLB1 基因,该基因在溶酶体β-半乳糖苷酶和弹性蛋白结合蛋白(EBP)中都有,导致 EBP 的弹性蛋白结合域缺失。
在汉族患者中发现的所有四个突变均导致β-半乳糖苷酶活性显著抑制,与疾病严重程度和是否伴有心肌病相关。