Department of Biomedical Informatics, Peking University Health Science Center, Beijing, China.
PLoS One. 2010 Nov 15;5(11):e15411. doi: 10.1371/journal.pone.0015411.
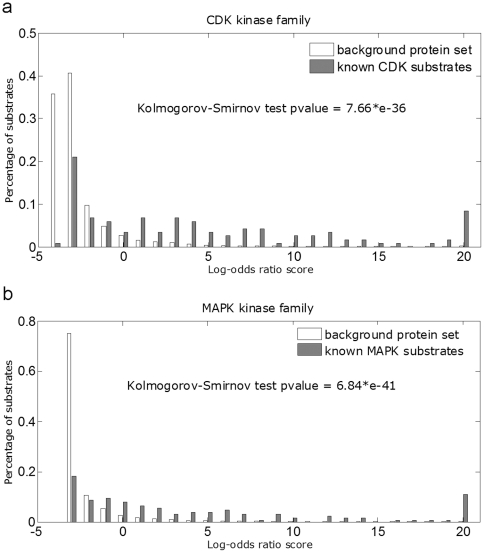
Phosphorylation is an important type of protein post-translational modification. Identification of possible phosphorylation sites of a protein is important for understanding its functions. Unbiased screening for phosphorylation sites by in vitro or in vivo experiments is time consuming and expensive; in silico prediction can provide functional candidates and help narrow down the experimental efforts. Most of the existing prediction algorithms take only the polypeptide sequence around the phosphorylation sites into consideration. However, protein phosphorylation is a very complex biological process in vivo. The polypeptide sequences around the potential sites are not sufficient to determine the phosphorylation status of those residues. In the current work, we integrated various data sources such as protein functional domains, protein subcellular location and protein-protein interactions, along with the polypeptide sequences to predict protein phosphorylation sites. The heterogeneous information significantly boosted the prediction accuracy for some kinase families. To demonstrate potential application of our method, we scanned a set of human proteins and predicted putative phosphorylation sites for Cyclin-dependent kinases, Casein kinase 2, Glycogen synthase kinase 3, Mitogen-activated protein kinases, protein kinase A, and protein kinase C families (available at http://cmbi.bjmu.edu.cn/huphospho). The predicted phosphorylation sites can serve as candidates for further experimental validation. Our strategy may also be applicable for the in silico identification of other post-translational modification substrates.
磷酸化是蛋白质翻译后修饰的一种重要类型。鉴定蛋白质的潜在磷酸化位点对于理解其功能非常重要。通过体外或体内实验进行无偏筛选磷酸化位点既耗时又昂贵;而基于计算的预测可以提供功能候选物,并有助于缩小实验工作的范围。大多数现有的预测算法仅考虑磷酸化位点周围的多肽序列。然而,蛋白质磷酸化是体内一个非常复杂的生物过程。潜在位点周围的多肽序列不足以确定这些残基的磷酸化状态。在目前的工作中,我们整合了各种数据源,如蛋白质功能域、蛋白质亚细胞定位和蛋白质-蛋白质相互作用,以及多肽序列,以预测蛋白质磷酸化位点。这些异构信息显著提高了一些激酶家族的预测准确性。为了展示我们方法的潜在应用,我们扫描了一组人类蛋白质,并预测了细胞周期蛋白依赖性激酶、酪蛋白激酶 2、糖原合成酶激酶 3、丝裂原活化蛋白激酶、蛋白激酶 A 和蛋白激酶 C 家族的潜在磷酸化位点(可在 http://cmbi.bjmu.edu.cn/huphospho 获得)。预测的磷酸化位点可以作为进一步实验验证的候选物。我们的策略也可能适用于其他翻译后修饰底物的计算鉴定。