Institute of Bioinformatics and Systems Biology, National Chiao Tung University, Hsinchu 300, Taiwan.
Nucleic Acids Res. 2011 Mar;39(5):e28. doi: 10.1093/nar/gkq1249. Epub 2010 Dec 3.
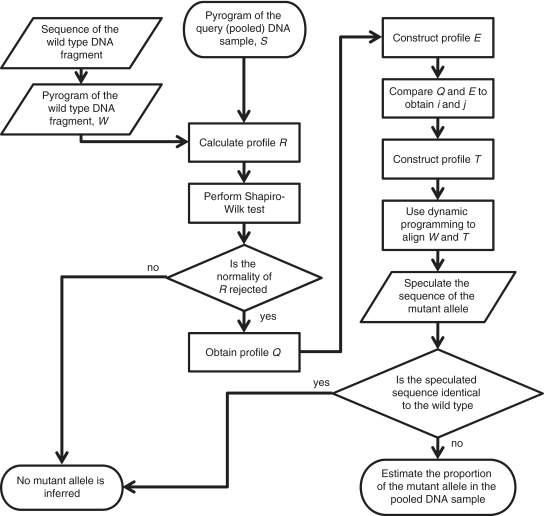
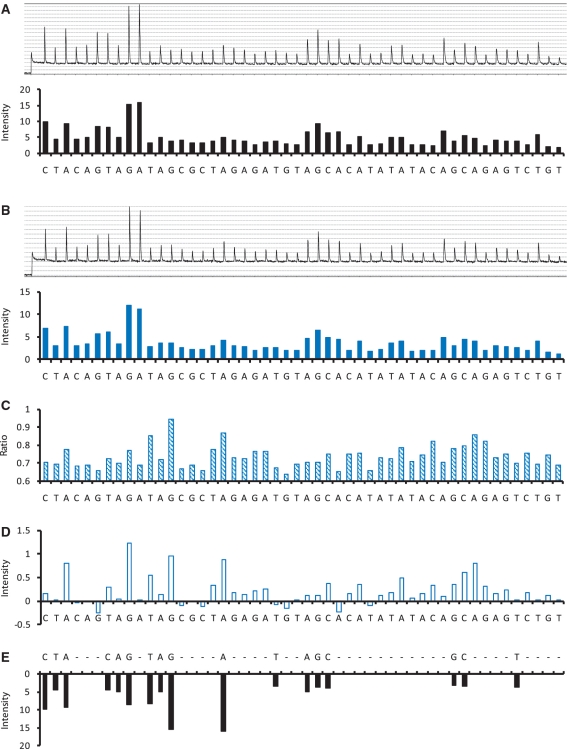
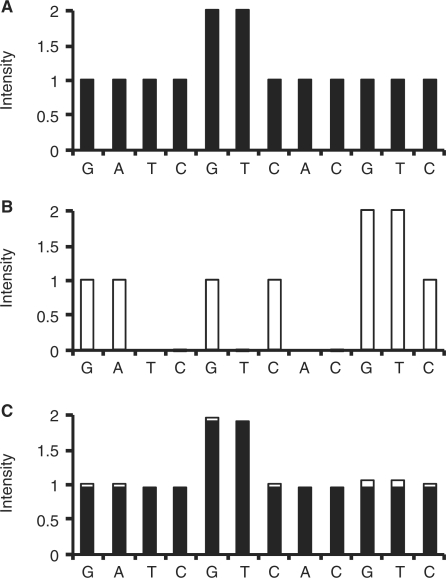
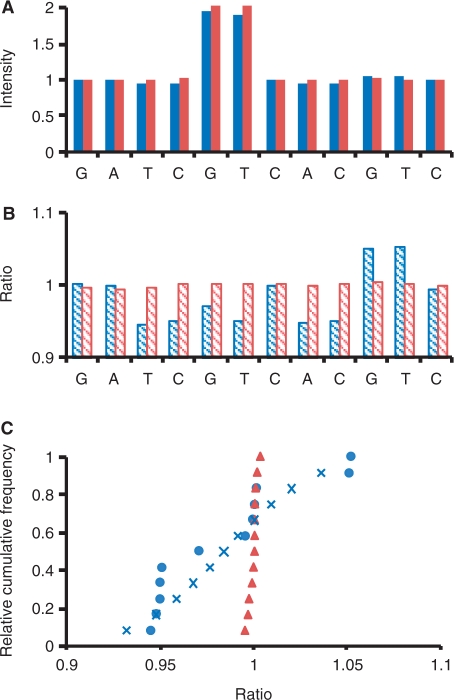
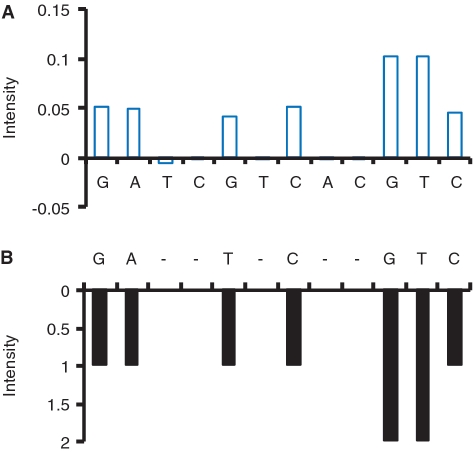
A practical way to reduce the cost of surveying single-nucleotide polymorphism (SNP) in a large number of individuals is to measure the allele frequencies in pooled DNA samples. Pyrosequencing(TM) has been frequently used for this application because signals generated by this approach are proportional to the amount of DNA templates. The Pyrosequencing(TM) pyrogram is determined by the dispensing order of dNTPs, which is usually designed based on the known SNPs to avoid asynchronistic extensions of heterozygous sequences. Therefore, utilizing the pyrogram signals to identify de novo SNPs in DNA pools has never been undertook. Here, in this study we developed an algorithm to address this issue. With the sequence and pyrogram of the wild-type allele known in advance, we could use the pyrogram obtained from the pooled DNA sample to predict the sequence of the unknown mutant allele (de novo SNP) and estimate its allele frequency. Both computational simulation and experimental Pyrosequencing(TM) test results suggested that our method performs well. The web interface of our method is available at http://life.nctu.edu.tw/∼yslin/PSM/.
在大量个体中降低单核苷酸多态性(SNP)检测成本的一种实用方法是测量混合 DNA 样本中的等位基因频率。焦磷酸测序(TM)经常用于这种应用,因为这种方法产生的信号与 DNA 模板的数量成正比。焦磷酸测序(TM)的焦谱图由 dNTP 的分配顺序决定,通常根据已知的 SNPs 进行设计,以避免异源序列的异步延伸。因此,从未利用焦谱图信号来识别 DNA 池中的新出现的 SNPs。在这里,在本研究中我们开发了一种算法来解决这个问题。通过预先知道野生型等位基因的序列和焦谱图,我们可以使用从混合 DNA 样本中获得的焦谱图来预测未知突变等位基因(新出现的 SNP)的序列,并估计其等位基因频率。计算模拟和实验焦磷酸测序(TM)测试结果均表明我们的方法表现良好。我们方法的网络界面可在 http://life.nctu.edu.tw/∼yslin/PSM/ 获得。