Department of Medicine, The University of Illinois at Chicago, Chicago, Illinois, United States of America.
PLoS One. 2011 Jan 31;6(1):e16571. doi: 10.1371/journal.pone.0016571.
Earlier we have shown that extracellular sphingosine-1-phosphate (S1P) induces migration of human pulmonary artery endothelial cells (HPAECs) through the activation of S1P(1) receptor, PKCε, and PLD2-PKCζ-Rac1 signaling cascade. As endothelial cells generate intracellular S1P, here we have investigated the role of sphingosine kinases (SphKs) and S1P lyase (S1PL), that regulate intracellular S1P accumulation, in HPAEC motility.
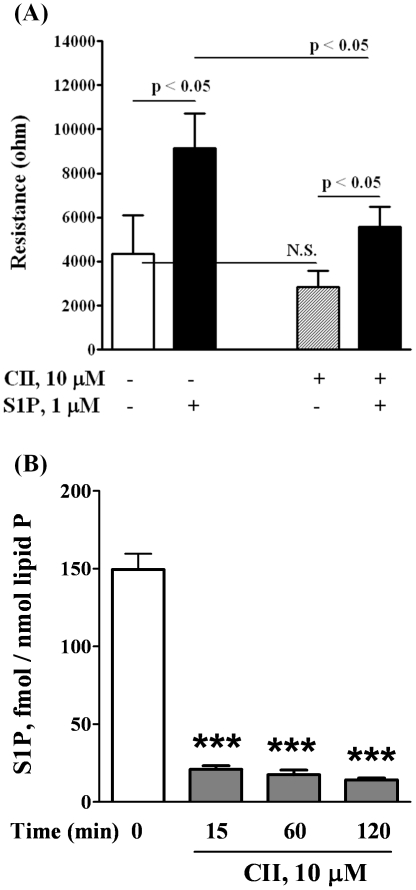
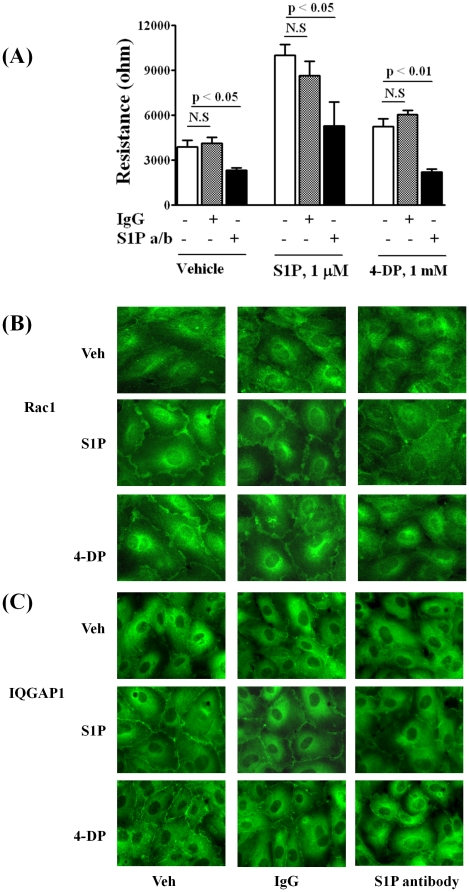
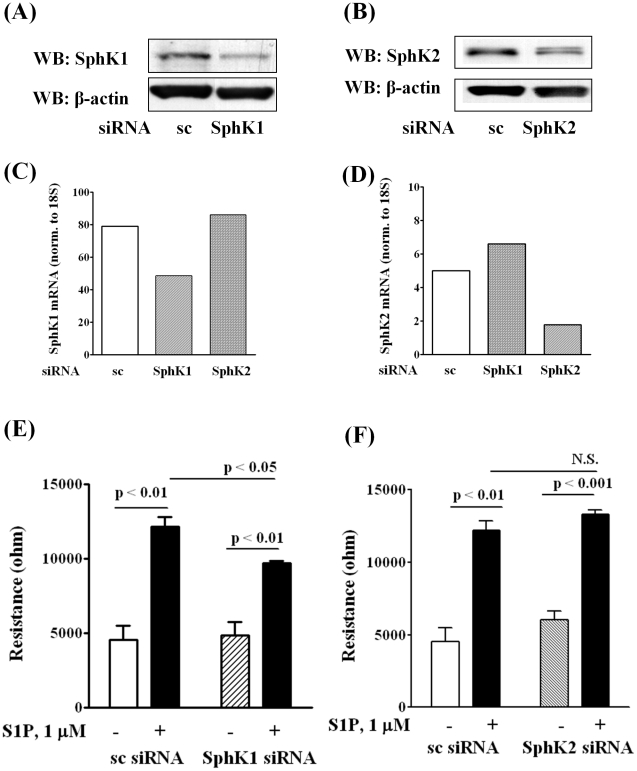
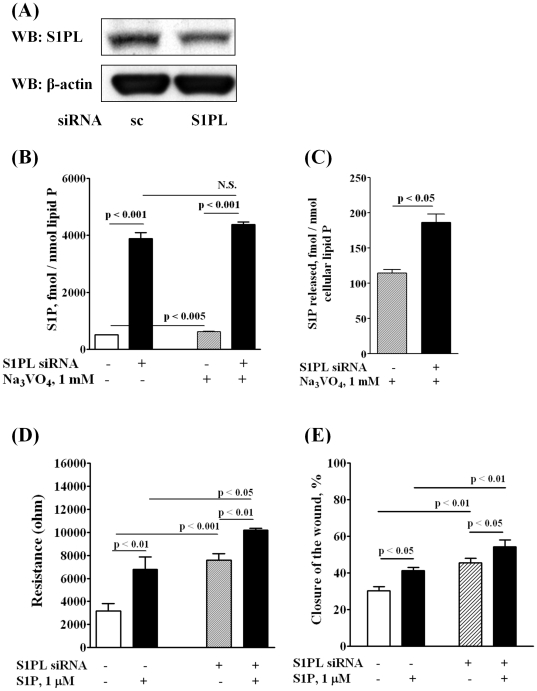
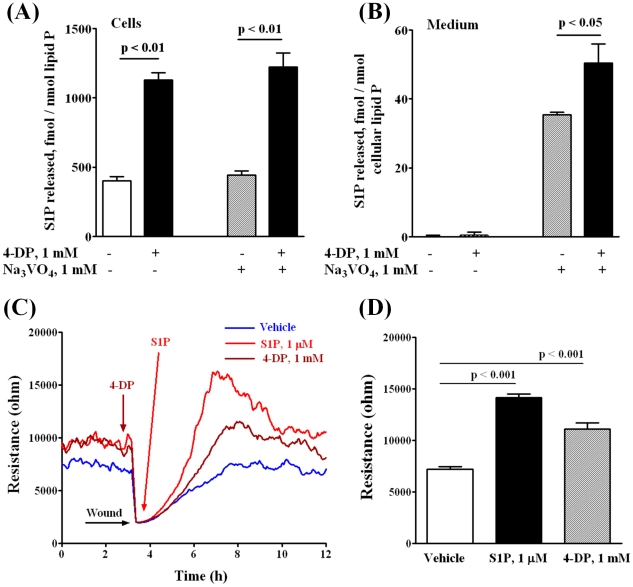
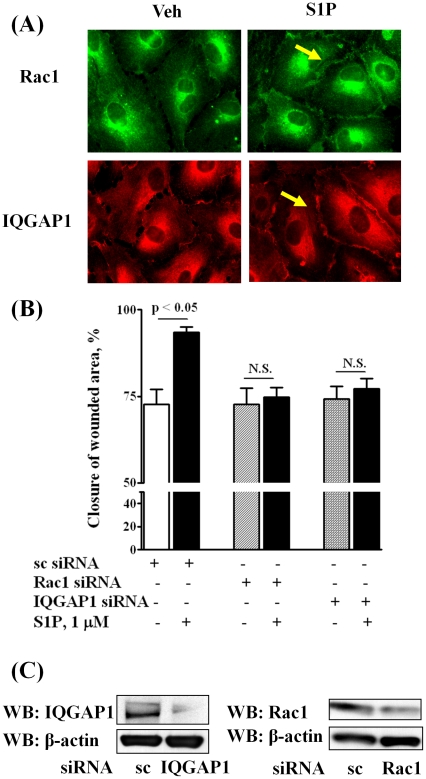
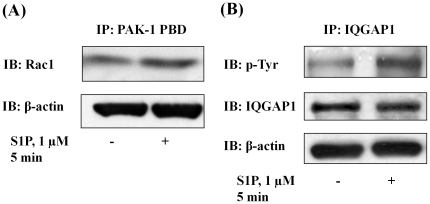
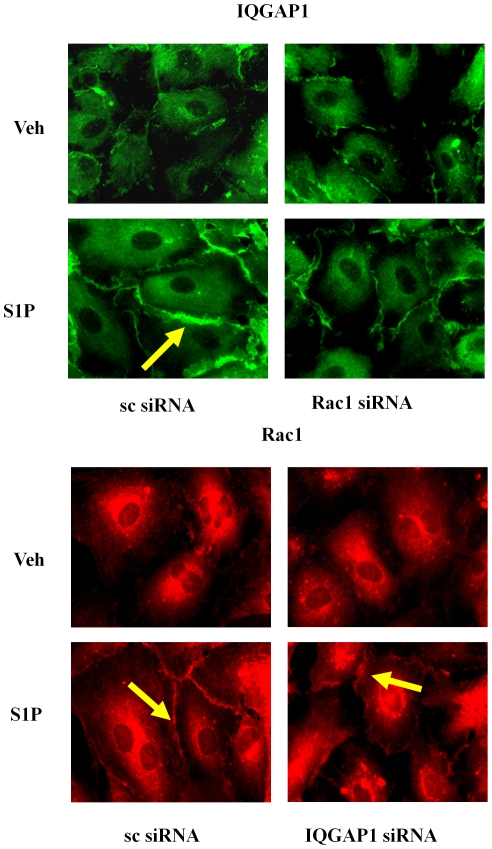
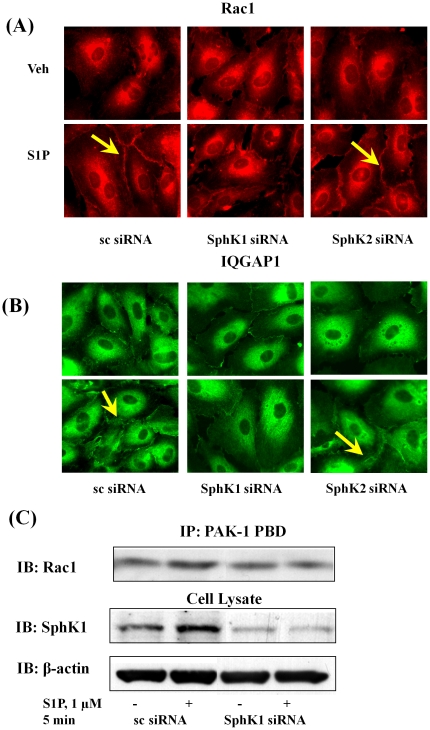
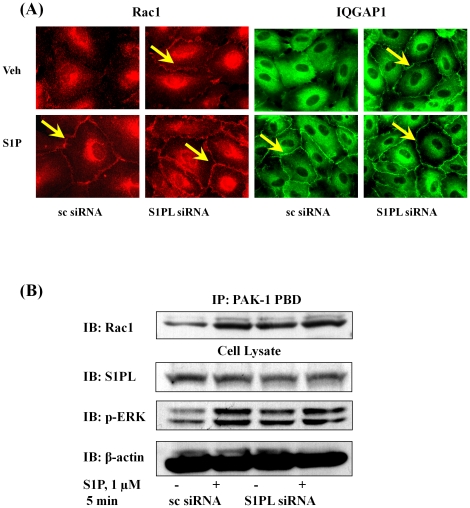
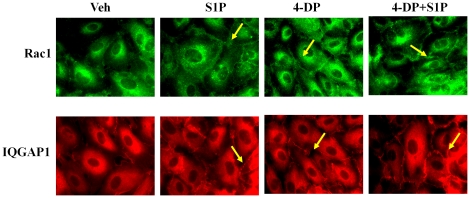
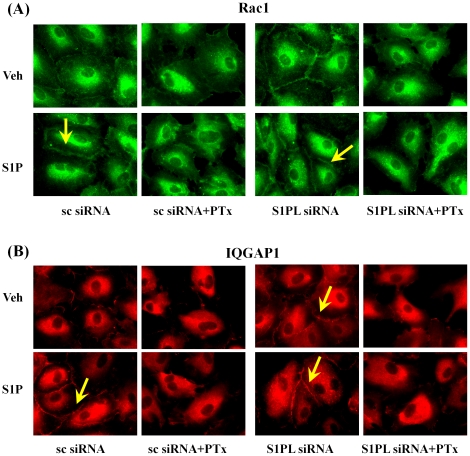
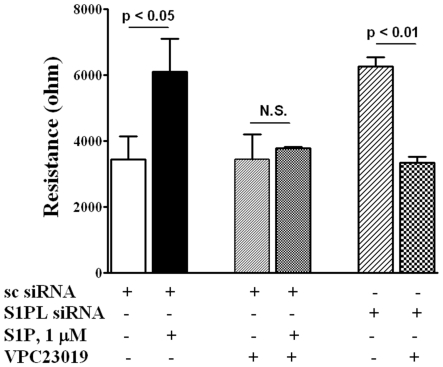
METHODOLOGY/PRINCIPAL FINDINGS: Inhibition of SphK activity with a SphK inhibitor 2-(p-Hydroxyanilino)-4-(p-Chlorophenyl) Thiazole or down-regulation of Sphk1, but not SphK2, with siRNA decreased S1P(int), and attenuated S1P(ext) or serum-induced motility of HPAECs. On the contrary, inhibition of S1PL with 4-deoxypyridoxine or knockdown of S1PL with siRNA increased S1P(int) and potentiated motility of HPAECs to S1P(ext) or serum. S1P(ext) mediates cell motility through activation of Rac1 and IQGAP1 signal transduction in HPAECs. Silencing of SphK1 by siRNA attenuated Rac1 and IQGAP1 translocation to the cell periphery; however, knockdown of S1PL with siRNA or 4-deoxypyridoxine augmented activated Rac1 and stimulated Rac1 and IQGAP1 translocation to cell periphery. The increased cell motility mediated by down-regulation was S1PL was pertussis toxin sensitive suggesting "inside-out" signaling of intracellularly generated S1P. Although S1P did not accumulate significantly in media under basal or S1PL knockdown conditions, addition of sodium vanadate increased S1P levels in the medium and inside the cells most likely by blocking phosphatases including lipid phosphate phosphatases (LPPs). Furthermore, addition of anti-S1P mAb to the incubation medium blocked S1P(ext) or 4-deoxypyridoxine-dependent endothelial cell motility.
CONCLUSIONS/SIGNIFICANCE: These results suggest S1P(ext) mediated endothelial cell motility is dependent on intracellular S1P production, which is regulated, in part, by SphK1 and S1PL.
我们之前已经表明,细胞外的鞘氨醇-1-磷酸(S1P)通过激活 S1P(1)受体、PKCε 和 PLD2-PKCζ-Rac1 信号级联诱导人肺动脉内皮细胞(HPAEC)迁移。由于内皮细胞产生细胞内 S1P,因此我们研究了调节细胞内 S1P 积累的鞘氨醇激酶(SphKs)和 S1P 裂解酶(S1PL)在 HPAEC 迁移中的作用。
方法/主要发现:用 SphK 抑制剂 2-(对-羟基苯胺基)-4-(对-氯苯基)噻唑或 siRNA 下调 Sphk1 抑制 SphK 活性,降低 S1P(int),并减弱 S1P(ext)或血清诱导的 HPAEC 迁移。相反,用 4-脱氧吡啶酮抑制 S1PL 或用 siRNA 敲低 S1PL 增加 S1P(int),增强 HPAEC 对 S1P(ext)或血清的迁移能力。S1P(ext)通过激活 Rac1 和 IQGAP1 信号转导在 HPAEC 中介导细胞迁移。siRNA 沉默 SphK1 减弱 Rac1 和 IQGAP1 向细胞边缘的易位;然而,siRNA 敲低 S1PL 或 4-脱氧吡啶酮增强了激活的 Rac1,并刺激 Rac1 和 IQGAP1 向细胞边缘易位。下调 S1PL 介导的细胞迁移是百日咳毒素敏感的,表明细胞内产生的 S1P 的“内-外”信号。尽管在基础或 S1PL 敲低条件下 S1P 在培养基中没有明显积累,但添加焦磷酸钠会增加培养基和细胞内的 S1P 水平,这很可能是通过阻断包括磷酸酶在内的磷酸酶,包括脂质磷酸酶(LPPs)。此外,将抗 S1P mAb 添加到孵育培养基中会阻断 S1P(ext)或 4-脱氧吡啶酮依赖性内皮细胞迁移。
结论/意义:这些结果表明,S1P(ext)介导的内皮细胞迁移依赖于细胞内 S1P 的产生,这部分受 SphK1 和 S1PL 的调节。