Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria 3052, Australia.
BMC Bioinformatics. 2011 Mar 8;12:68. doi: 10.1186/1471-2105-12-68.
Illumina's Infinium SNP BeadChips are extensively used in both small and large-scale genetic studies. A fundamental step in any analysis is the processing of raw allele A and allele B intensities from each SNP into genotype calls (AA, AB, BB). Various algorithms which make use of different statistical models are available for this task. We compare four methods (GenCall, Illuminus, GenoSNP and CRLMM) on data where the true genotypes are known in advance and data from a recently published genome-wide association study.
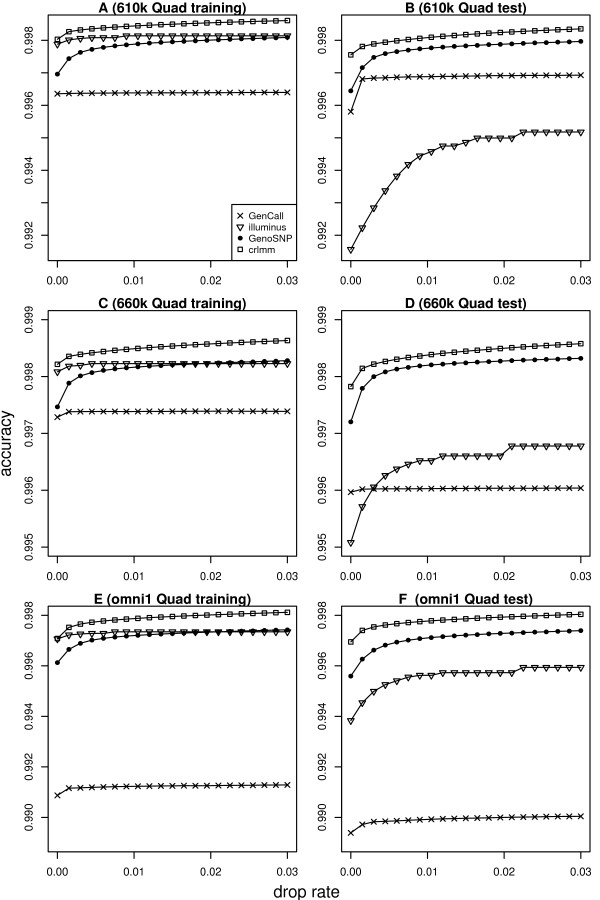
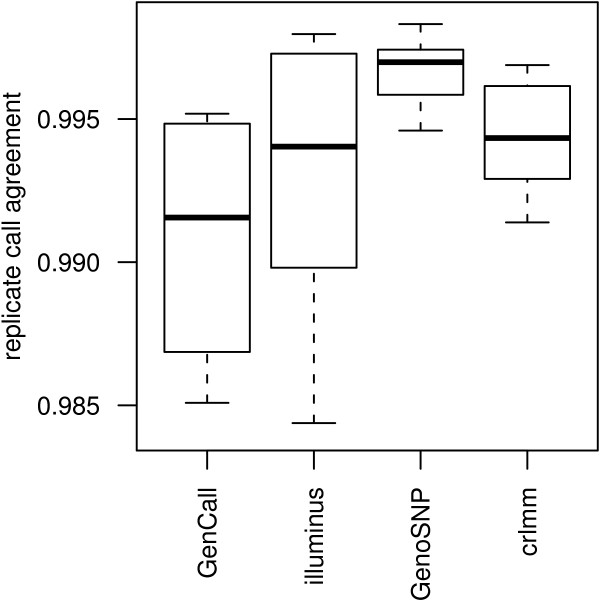
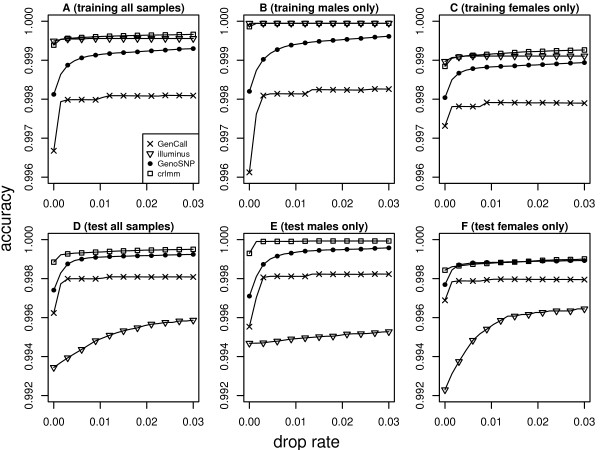
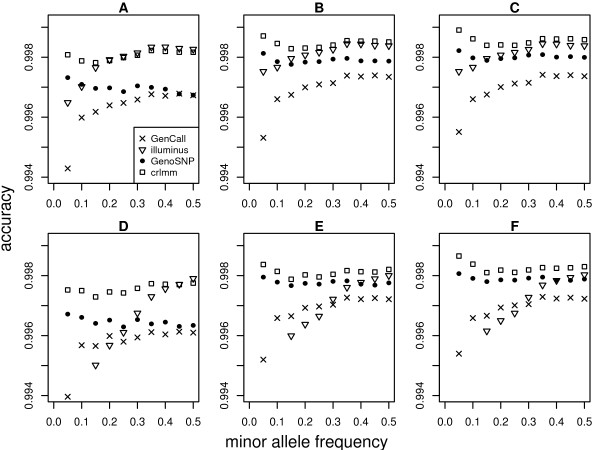
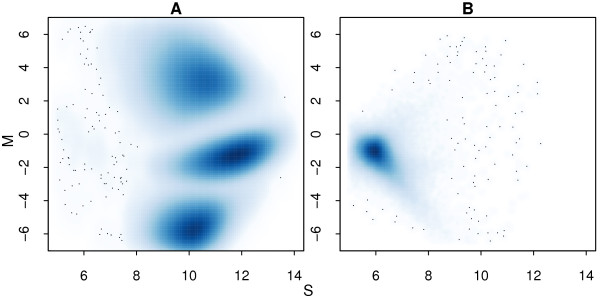
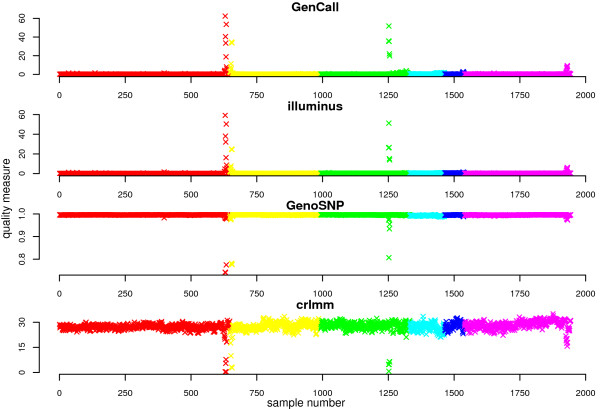
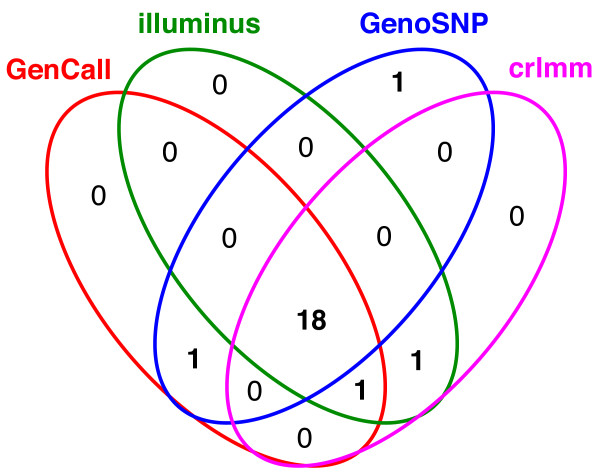
In general, differences in accuracy are relatively small between the methods evaluated, although CRLMM and GenoSNP were found to consistently outperform GenCall. The performance of Illuminus is heavily dependent on sample size, with lower no call rates and improved accuracy as the number of samples available increases. For X chromosome SNPs, methods with sex-dependent models (Illuminus, CRLMM) perform better than methods which ignore gender information (GenCall, GenoSNP). We observe that CRLMM and GenoSNP are more accurate at calling SNPs with low minor allele frequency than GenCall or Illuminus. The sample quality metrics from each of the four methods were found to have a high level of agreement at flagging samples with unusual signal characteristics.
CRLMM, GenoSNP and GenCall can be applied with confidence in studies of any size, as their performance was shown to be invariant to the number of samples available. Illuminus on the other hand requires a larger number of samples to achieve comparable levels of accuracy and its use in smaller studies (50 or fewer individuals) is not recommended.
Illumina 的 Infinium SNP BeadChips 广泛应用于小型和大型遗传研究中。任何分析的基本步骤都是将每个 SNP 的原始等位基因 A 和等位基因 B 强度处理为基因型调用(AA、AB、BB)。为此任务提供了各种利用不同统计模型的算法。我们比较了四种方法(GenCall、Illuminus、GenoSNP 和 CRLMM),一种方法是在已知真实基因型的数据上,另一种方法是在最近发表的全基因组关联研究的数据上。
一般来说,评估的方法之间准确性差异相对较小,尽管 CRLMM 和 GenoSNP 被发现始终优于 GenCall。Illuminus 的性能严重依赖于样本量,随着可用样本数量的增加,无调用率降低,准确性提高。对于 X 染色体 SNP,具有性别依赖模型的方法(Illuminus、CRLMM)比忽略性别信息的方法(GenCall、GenoSNP)表现更好。我们观察到 CRLMM 和 GenoSNP 在调用低次要等位基因频率 SNP 时比 GenCall 或 Illuminus 更准确。这四种方法中的每一种的样本质量指标在标记具有异常信号特征的样本方面具有高度一致性。
CRLMM、GenoSNP 和 GenCall 可以在任何规模的研究中自信地应用,因为它们的性能不受可用样本数量的影响。另一方面,Illuminus 需要更多的样本才能达到可比的准确性水平,不建议在较小的研究(50 或更少的个体)中使用。