Wang Meng, Zhang Jie, Zhou Jian-Hua, Chen Hao-Tai, Ma Li-Na, Ding Yao-Zhong, Liu Wen-Qian, Gu Yuan-Xing, Zhao Feng, Liu Yong-Sheng
State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou.
Biosystems. 2011 Oct;106(1):45-50. doi: 10.1016/j.biosystems.2011.06.005. Epub 2011 Jun 17.
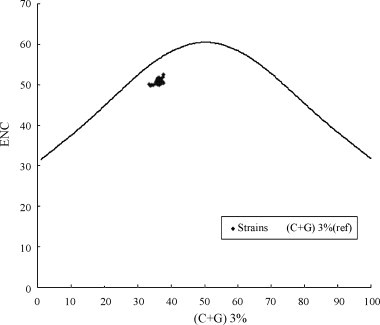
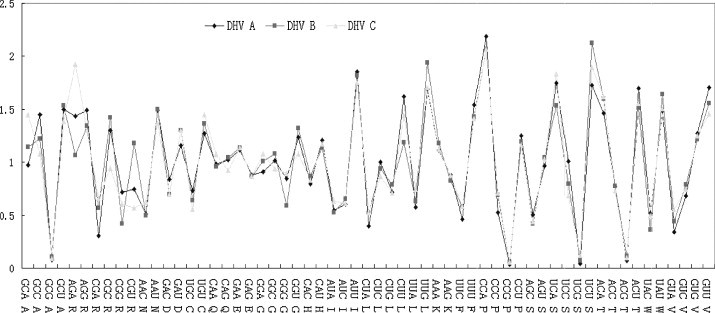
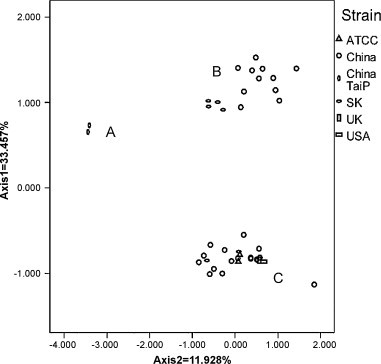
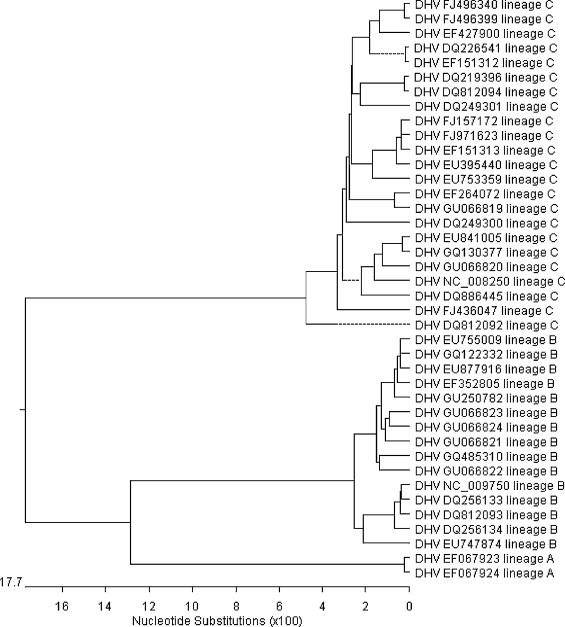
In this study, an abundant (A+U)% and low codon bias were revealed in duck hepatitis virus type 1 (DHV-1) and the new serotype strains isolated from Taiwan, South Korea and Mainland China (DHV-N). The general correlation between base composition and codon usage bias suggests that mutational pressure rather than natural selection is the main factor that determines the codon usage bias in these samples. By comparative analysis of the codon usage patterns of 40 ORFs of DHV, we found that all of DHV-1 strains grouped in genotype C; the DHV-N strains isolated in South Korea and China clustered into genotypes B; and the DHV-N strains isolated from Taiwan clustered into genotypes A. The findings revealed that more than one subtype of DHV-1 circulated in East Asia. Furthermore, the results of phylogenetic analyses based on RSCU values and Clustal W method indicated obvious phylogenetic congruities. This suggested that better genome consistency of DHV may exist in nature and phylogenetic analyses based on RSCU values maybe a good method in classifying genotypes of the virus. Our work might give some clues to the features and some evolutionary information of DHV.
在本研究中,1型鸭肝炎病毒(DHV-1)以及从台湾、韩国和中国大陆分离出的新血清型毒株(DHV-N)显示出较高的(A+U)%和较低的密码子偏好性。碱基组成与密码子使用偏好之间的一般相关性表明,突变压力而非自然选择是决定这些样本中密码子使用偏好的主要因素。通过对DHV的40个开放阅读框(ORF)的密码子使用模式进行比较分析,我们发现所有DHV-1毒株都归为C基因型;在韩国和中国分离出的DHV-N毒株聚为B基因型;从台湾分离出的DHV-N毒株聚为A基因型。这些发现表明,DHV-1的不止一种亚型在东亚地区传播。此外,基于相对同义密码子使用(RSCU)值和Clustal W方法的系统发育分析结果显示出明显的系统发育一致性。这表明DHV在自然界中可能存在更好的基因组一致性,基于RSCU值的系统发育分析可能是对该病毒进行基因型分类的一种好方法。我们的工作可能为DHV的特征和一些进化信息提供一些线索。