Human Genome Sequencing Center, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA.
Genome Biol. 2011 Jul 25;12(7):R68. doi: 10.1186/gb-2011-12-7-r68.
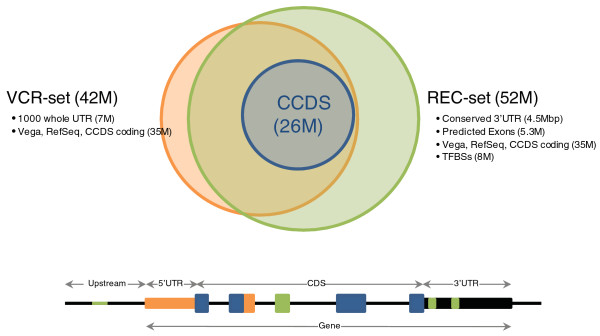
Enrichment of loci by DNA hybridization-capture, followed by high-throughput sequencing, is an important tool in modern genetics. Currently, the most common targets for enrichment are the protein coding exons represented by the consensus coding DNA sequence (CCDS). The CCDS, however, excludes many actual or computationally predicted coding exons present in other databases, such as RefSeq and Vega, and non-coding functional elements such as untranslated and regulatory regions. The number of variants per base pair (variant density) and our ability to interrogate regions outside of the CCDS regions is consequently less well understood.
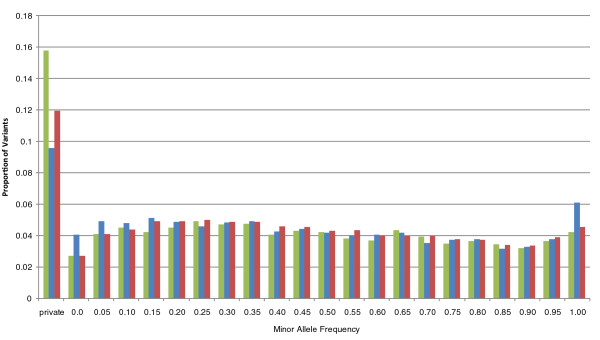
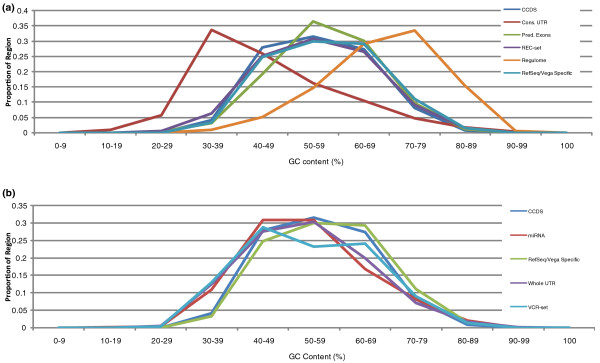
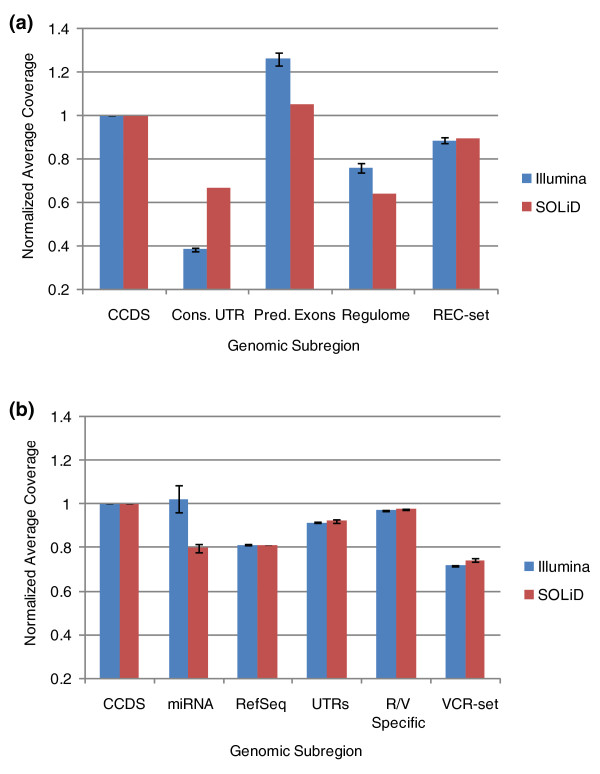
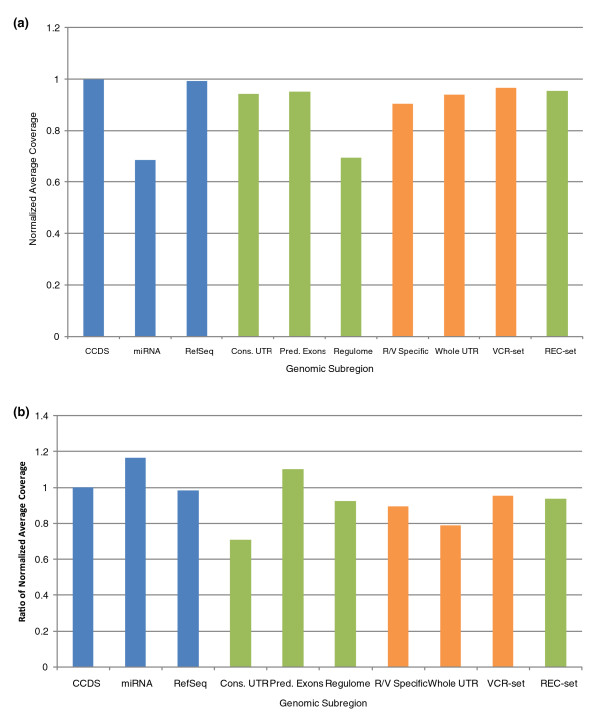
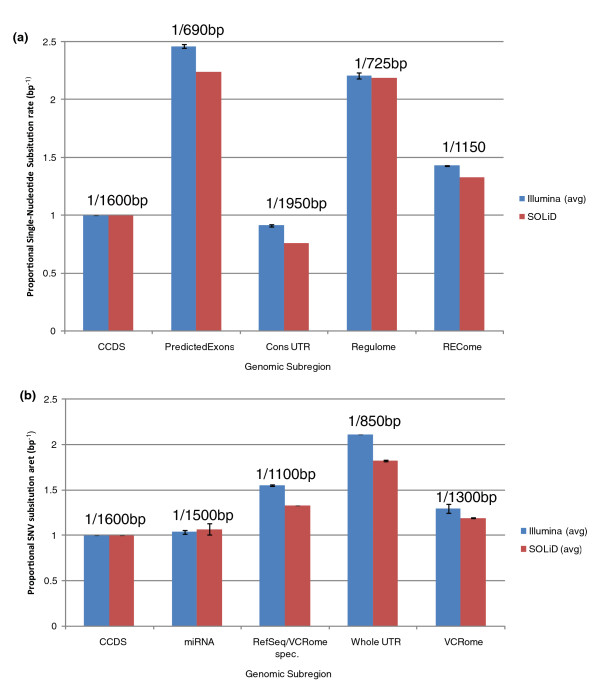
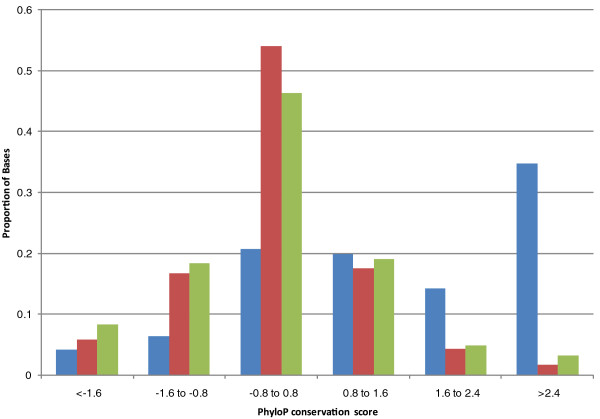
We examine capture sequence data from outside of the CCDS regions and find that extremes of GC content that are present in different subregions of the genome can reduce the local capture sequence coverage to less than 50% relative to the CCDS. This effect is due to biases inherent in both the Illumina and SOLiD sequencing platforms that are exacerbated by the capture process. Interestingly, for two subregion types, microRNA and predicted exons, the capture process yields higher than expected coverage when compared to whole genome sequencing. Lastly, we examine the variation present in non-CCDS regions and find that predicted exons, as well as exonic regions specific to RefSeq and Vega, show much higher variant densities than the CCDS.
We show that regions outside of the CCDS perform less efficiently in capture sequence experiments. Further, we show that the variant density in computationally predicted exons is more than 2.5-times higher than that observed in the CCDS.
通过 DNA 杂交捕获富集,然后进行高通量测序,是现代遗传学中的重要工具。目前,富集的最常见目标是由共识编码 DNA 序列(CCDS)代表的蛋白编码外显子。然而,CCDS 排除了许多实际存在或通过计算预测存在于其他数据库(如 RefSeq 和 Vega)中的编码外显子,以及非编码功能元件,如非翻译和调节区域。每个碱基对的变异数量(变异密度)以及我们在外显子区域外进行检测的能力因此了解得不够充分。
我们检查了 CCDS 区域外的捕获序列数据,发现基因组不同亚区存在的极端 GC 含量会使局部捕获序列覆盖率相对于 CCDS 降低到 50%以下。这种效应是由于 Illumina 和 SOLiD 测序平台固有的偏倚,再加上捕获过程的影响而加剧。有趣的是,对于 microRNA 和预测外显子这两种亚区类型,与全基因组测序相比,捕获过程产生的覆盖度高于预期。最后,我们检查了非 CCDS 区域的变异情况,发现与 CCDS 相比,预测外显子以及 RefSeq 和 Vega 特有的外显子区域的变异密度要高得多。
我们表明 CCDS 区域外的区域在捕获序列实验中的表现效率较低。此外,我们还表明,计算预测外显子中的变异密度比 CCDS 中观察到的高出 2.5 倍以上。