Howard Hughes Medical Institute, Department of Biochemistry and Molecular Biophysics, Columbia University, New York, New York, United States of America.
PLoS Comput Biol. 2011 Oct;7(10):e1002175. doi: 10.1371/journal.pcbi.1002175. Epub 2011 Oct 6.
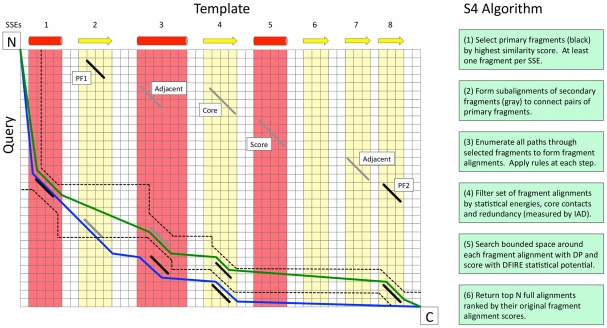
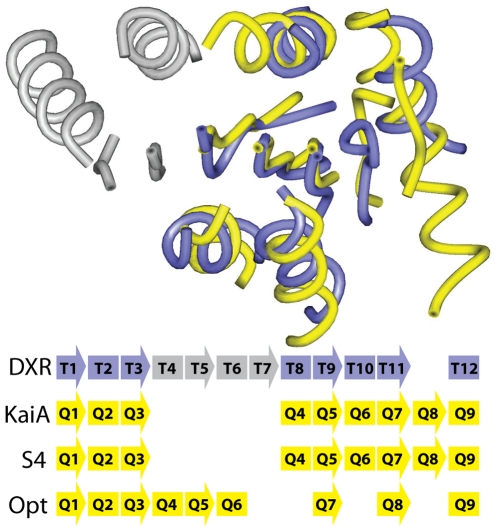
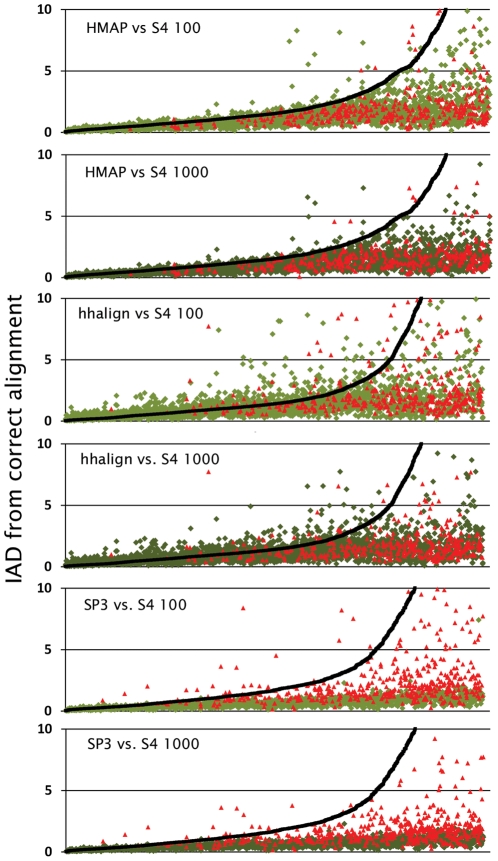
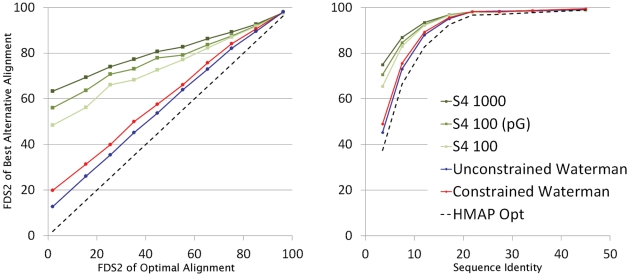
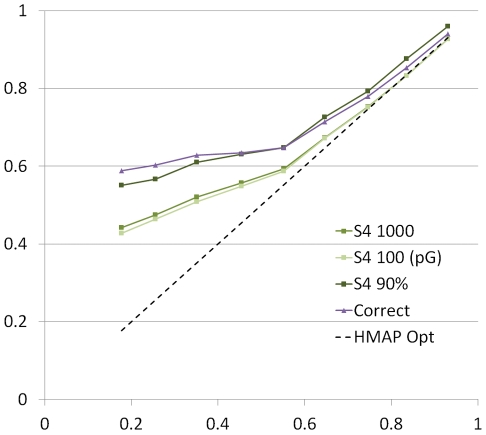
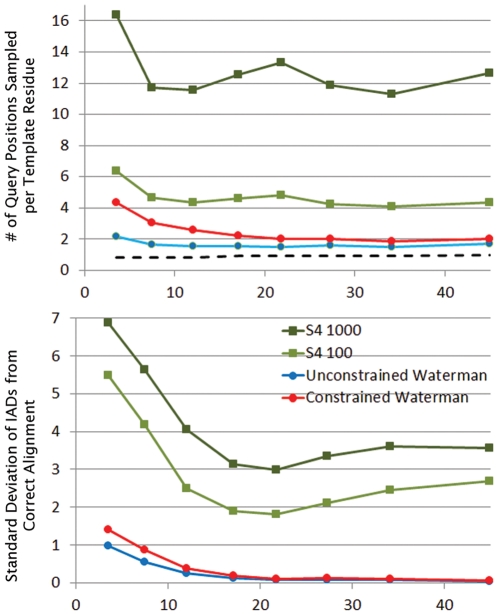
Protein structure modeling by homology requires an accurate sequence alignment between the query protein and its structural template. However, sequence alignment methods based on dynamic programming (DP) are typically unable to generate accurate alignments for remote sequence homologs, thus limiting the applicability of modeling methods. A central problem is that the alignment that is "optimal" in terms of the DP score does not necessarily correspond to the alignment that produces the most accurate structural model. That is, the correct alignment based on structural superposition will generally have a lower score than the optimal alignment obtained from sequence. Variations of the DP algorithm have been developed that generate alternative alignments that are "suboptimal" in terms of the DP score, but these still encounter difficulties in detecting the correct structural alignment. We present here a new alternative sequence alignment method that relies heavily on the structure of the template. By initially aligning the query sequence to individual fragments in secondary structure elements and combining high-scoring fragments that pass basic tests for "modelability", we can generate accurate alignments within a small ensemble. Our results suggest that the set of sequences that can currently be modeled by homology can be greatly extended.
同源蛋白结构建模需要在查询蛋白与其结构模板之间进行精确的序列比对。然而,基于动态规划 (DP) 的序列比对方法通常无法为远程序列同源物生成准确的比对,从而限制了建模方法的适用性。一个核心问题是,根据 DP 得分“最优”的比对并不一定对应于生成最准确结构模型的比对。也就是说,基于结构叠加的正确比对通常比从序列获得的最优比对得分低。已经开发了 DP 算法的变体,这些变体生成了在 DP 得分方面“次优”的替代比对,但在检测正确的结构比对时仍然存在困难。我们在这里提出了一种新的替代序列比对方法,该方法严重依赖于模板的结构。通过最初将查询序列与二级结构元素中的各个片段对齐,并组合通过“可建模性”基本测试的高得分片段,我们可以在小集合内生成准确的比对。我们的结果表明,目前可以通过同源建模的序列集可以大大扩展。