Molecular Investigation of Genetic Orphan Diseases, Pasteur Institute, Tunis - Tunisia.
Diagn Pathol. 2012 Jan 10;7:4. doi: 10.1186/1746-1596-7-4.
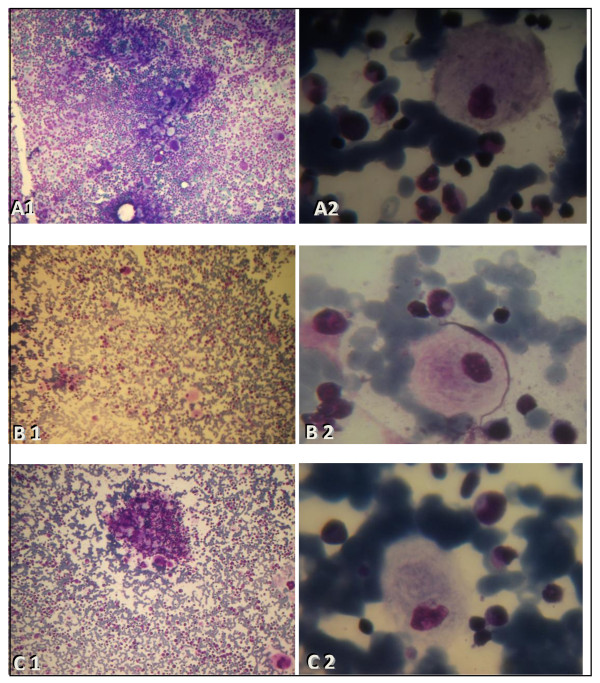
Gaucher disease (GD) is the most frequent lysosomal storage disorder; type 1 is by far the most common form. It is characterized by variability in age of onset, clinical signs and progression. It is usually diagnosed in the first or second decade of life with the appearance of bone pains, splenomegaly and thrombocytopenia, but the disease may be diagnosed at any age between 1 and 73 years. In the present study, we report 3 cases with late onset of GD in whom the disease was a surprise finding including one patient with Parkinson disease. This late onset is described as an adult form of Gaucher disease.
Molecular investigation showed mutational homogeneity in Tunisian adult patients suffering from GD. Indeed, all patients carry the p.N370S mutation: two patients at a homozygous state and one patient at compound heterozygous state.
The p.N370S mutation presents a large variability in the onset of the disease and its clinical manifestation supporting the view that GD should be considered as a continuum phenotype rather than a predefined classification.
戈谢病(Gaucher disease,GD)是最常见的溶酶体贮积症;1 型是迄今为止最常见的形式。其发病年龄、临床表现和进展各不相同。它通常在生命的第一个或第二个十年被诊断出来,表现为骨痛、脾肿大和血小板减少症,但该疾病也可能在 1 至 73 岁之间的任何年龄被诊断出来。在本研究中,我们报告了 3 例晚发型 GD 病例,其中疾病的发现令人意外,包括 1 例帕金森病患者。这种晚发型被描述为成人型戈谢病。
分子研究显示突尼斯成年 GD 患者具有突变同源性。事实上,所有患者均携带 p.N370S 突变:2 例为纯合子状态,1 例为复合杂合子状态。
p.N370S 突变在疾病的发病和临床表现上具有很大的变异性,支持将 GD 视为连续表型而不是预先定义的分类的观点。