Radiopharmaceutical Chemistry Laboratory, Turku PET Centre, University of Turku, Porthaninkatu 3, Turku, 20500, Finland.
EJNMMI Res. 2012 Jan 25;2(1):3. doi: 10.1186/2191-219X-2-3.
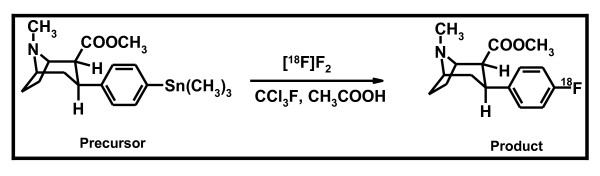
We present the electrophilic synthesis of [18F]2β-carbomethoxy-3β-(4-fluoro)tropane [[18F]CFT] and the pharmacological specificity and selectivity of [18F]CFT for monoamine transporters in the brain and peripheral organs of rats. The human radiation dose is extrapolated from the animal data.
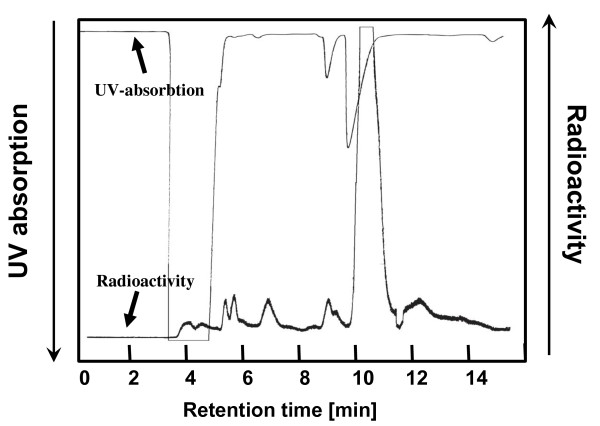
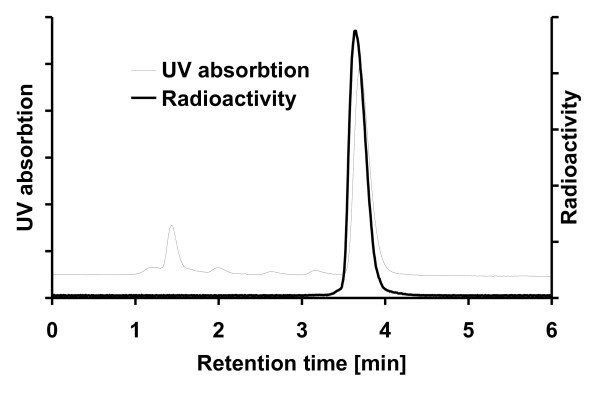
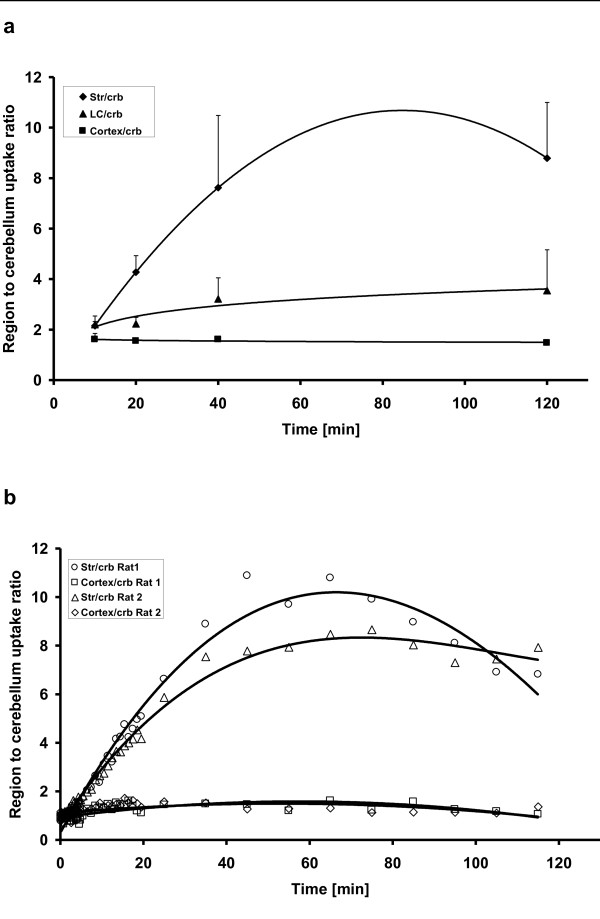
[18F]CFT was synthesized by electrophilic fluorination of a stannylated precursor by using post-target-produced [18F]F2 as a fluorinating agent. The ex vivo 18F-activity biodistribution of [18F]CFT in the brain of rats was studied by autoradiography. The binding of [18F]CFT to the monoamine transporters was studied using in vivo blocking experiments with dopamine transporter [DAT], norepinephrine transporter [NET], or serotonin transporter [SERT] inhibitors. In vivo animal positron emission tomography was used as a comparative method to determine tracer kinetics. Human radiation dose was assessed using OLINDA software.
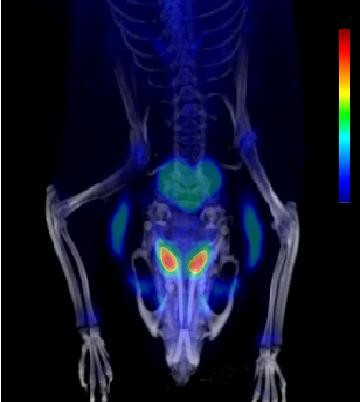
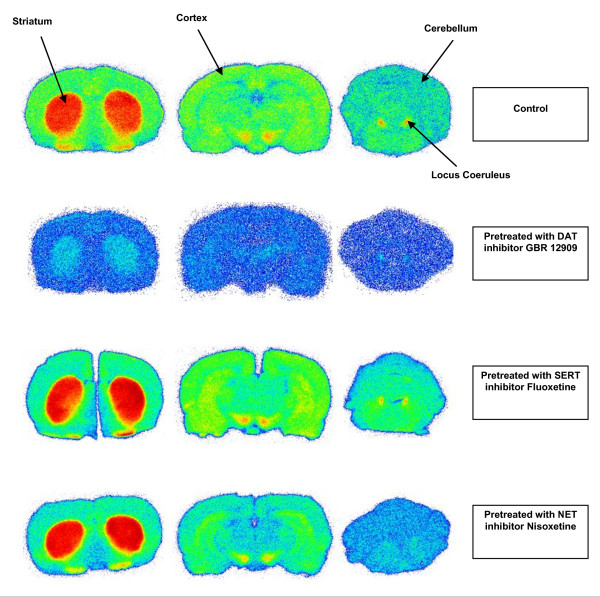
The radiochemical yield of [18F]CFT from the initial [18F]F-, decay corrected to the end of bombardment, was 3.2 ± 1.0%. The specific activity [SA] was 14.5 ± 3.4 GBq/μmol, decay corrected to the end of synthesis. Radiochemical purity exceeded 99%. DAT-specific binding was found in the striatum, locus coeruleus, and pancreas. NET-specific binding was found in the locus coeruleus. SERT-specific binding was not found in any of the studied organs. Effective dose equivalent [EDE] estimated for the standard human model was 12.8 μSv/MBq. Effective dose [ED] was 9.17 μSv/MBq.
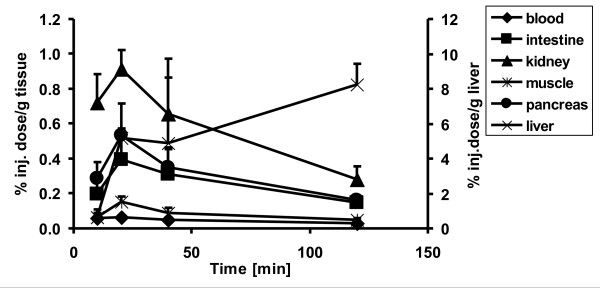
Post-target-produced high-SA [18F]F2 was used to incorporate18F directly into the phenyl ring of [18F]CFT. The final product had high radiochemical and chemical purities and a high SA for DAT and NET studies in vivo. In periphery, [18F]CFT showed a specific uptake in the pancreas. EDE and ED corresponded well with other18F-radioligands.
我们介绍了[18F]2β- 羧甲基-3β-(4-氟)托烷 [[18F]CFT] 的亲电合成,以及[18F]CFT 在大鼠脑和外周器官中单胺转运体的药理学特异性和选择性。人体辐射剂量是从动物数据推断出来的。
通过使用后靶产生的[18F]F2 作为氟源,对锡化前体进行亲电氟化,合成[18F]CFT。通过放射自显影研究[18F]CFT 在大鼠脑内的体外 18F- 活性生物分布。使用多巴胺转运体 [DAT]、去甲肾上腺素转运体 [NET] 或 5-羟色胺转运体 [SERT] 抑制剂的体内阻断实验研究[18F]CFT 与单胺转运体的结合。体内动物正电子发射断层扫描(PET)被用作比较方法来确定示踪剂动力学。使用 OLINDA 软件评估人体辐射剂量。
从初始[18F]F-,衰减校正至末端的放射性化学产率为 3.2±1.0%。比活度[SA]为 14.5±3.4GBq/μmol,衰减校正至合成结束。放射化学纯度超过 99%。DAT 特异性结合在纹状体、蓝斑核和胰腺中被发现。NET 特异性结合在蓝斑核中被发现。在研究的任何器官中均未发现 SERT 特异性结合。标准人体模型估计的有效剂量当量[EDE]为 12.8μSv/MBq。有效剂量[ED]为 9.17μSv/MBq。
使用后靶产生的高 SA [18F]F2 将 18F 直接掺入 [18F]CFT 的苯环中。最终产物具有高放射化学和化学纯度,以及用于体内 DAT 和 NET 研究的高 SA。在外周,[18F]CFT 在胰腺中表现出特异性摄取。EDE 和 ED 与其他 18F 放射性配体非常吻合。