MicroDiscovery GmbH, Marienburger Str, 1, 10405 Berlin, Germany.
BMC Bioinformatics. 2012 Feb 16;13:34. doi: 10.1186/1471-2105-13-34.
Recent development of novel technologies paved the way for quantitative proteomics. One of the most important among them is iTRAQ, employing isobaric tags for relative or absolute quantitation. Despite large progress in technology development, still many challenges remain for derivation and interpretation of quantitative results. One of these challenges is the consistent assignment of peptides to proteins.
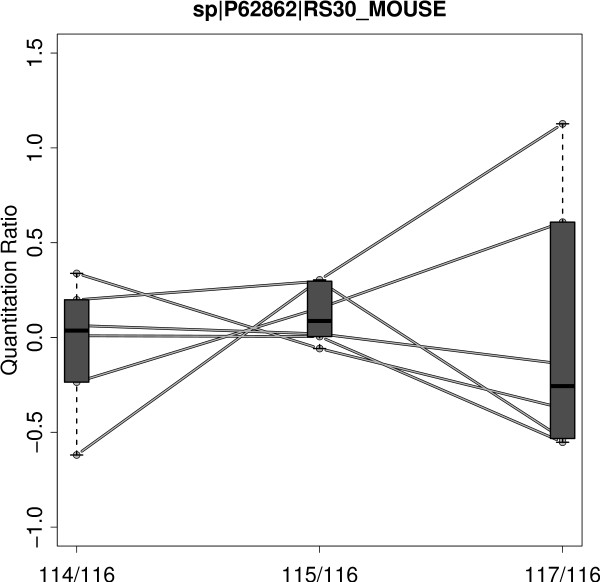
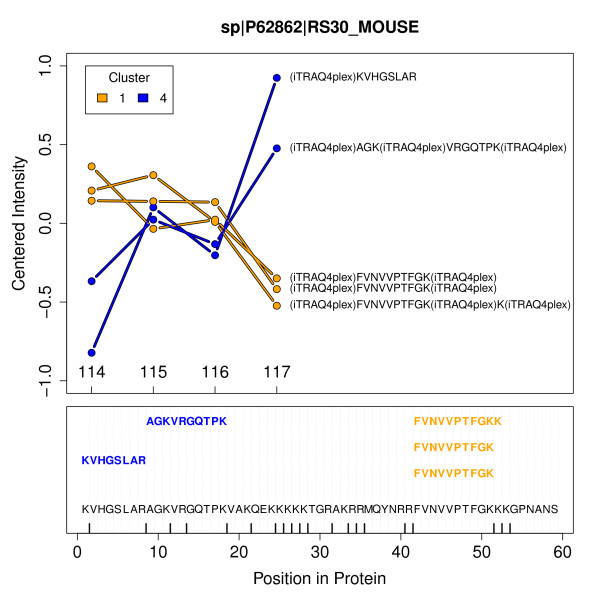
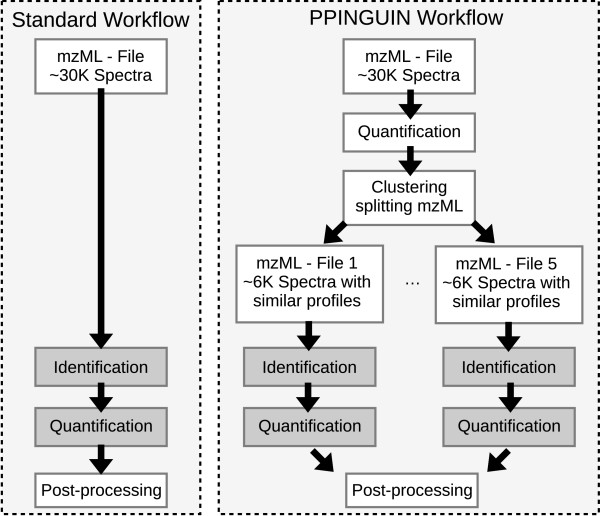
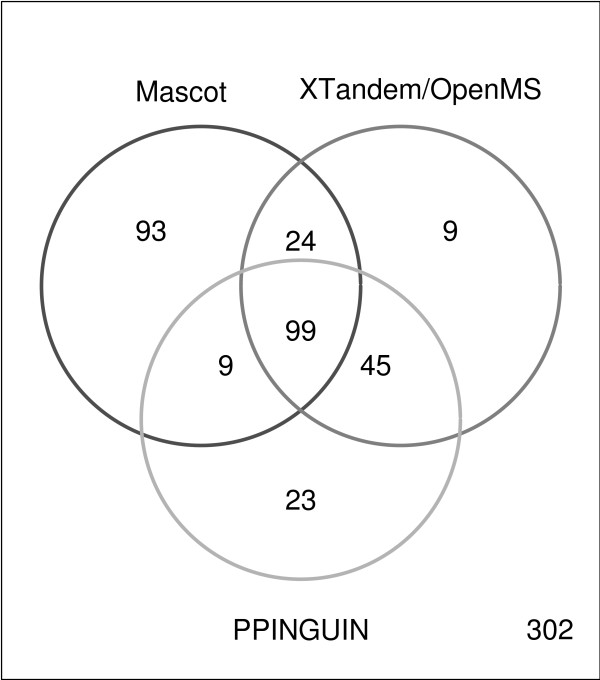
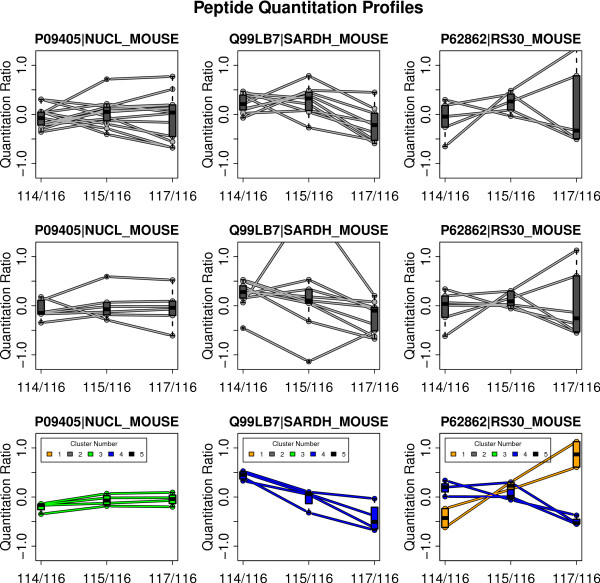
We have developed Peptide Profiling Guided Identification of Proteins (PPINGUIN), a statistical analysis workflow for iTRAQ data addressing the problem of ambiguous peptide quantitations. Motivated by the assumption that peptides uniquely derived from the same protein are correlated, our method employs clustering as a very early step in data processing prior to protein inference. Our method increases experimental reproducibility and decreases variability of quantitations of peptides assigned to the same protein. Giving further support to our method, application to a type 2 diabetes dataset identifies a list of protein candidates that is in very good agreement with previously performed transcriptomics meta analysis. Making use of quantitative properties of signal patterns identified, PPINGUIN can reveal new isoform candidates.
Regarding the increasing importance of quantitative proteomics we think that this method will be useful in practical applications like model fitting or functional enrichment analysis. We recommend to use this method if quantitation is a major objective of research.
新型技术的最新发展为定量蛋白质组学铺平了道路。其中最重要的技术之一是 iTRAQ,它采用同量异位标签进行相对或绝对定量。尽管在技术开发方面取得了很大进展,但在推导和解释定量结果方面仍存在许多挑战。其中一个挑战是将肽一致地分配到蛋白质中。
我们开发了肽谱引导蛋白鉴定(PPINGUIN),这是一种针对 iTRAQ 数据的统计分析工作流程,用于解决定量肽不明确的问题。受从同一蛋白质中独特衍生的肽相关的假设的启发,我们的方法在蛋白质推断之前的非常早期数据处理步骤中使用聚类。我们的方法提高了实验的可重复性,并降低了分配给同一蛋白质的肽的定量变异性。将其应用于 2 型糖尿病数据集进一步支持了我们的方法,该方法确定了一组蛋白质候选物,与之前进行的转录组学荟萃分析非常吻合。利用识别出的信号模式的定量特性,PPINGUIN 可以揭示新的同工型候选物。
鉴于定量蛋白质组学的重要性日益增加,我们认为这种方法在模型拟合或功能富集分析等实际应用中会很有用。如果定量是研究的主要目标,我们建议使用此方法。