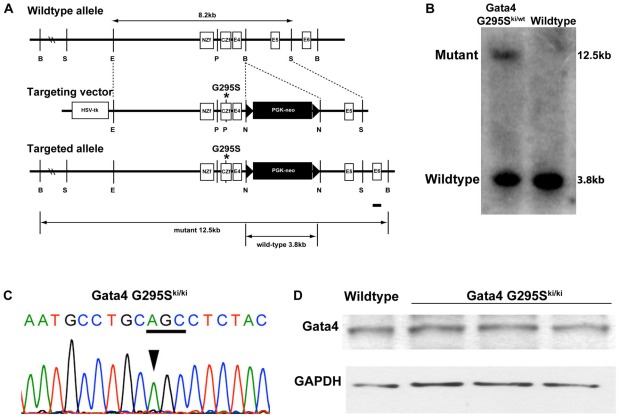
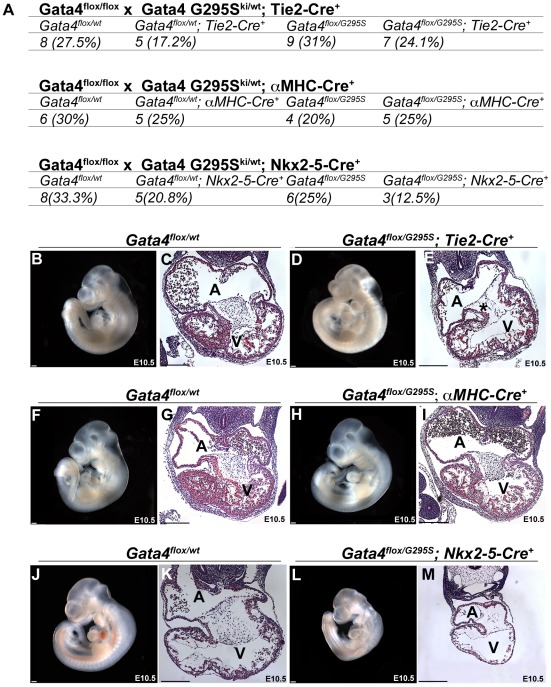
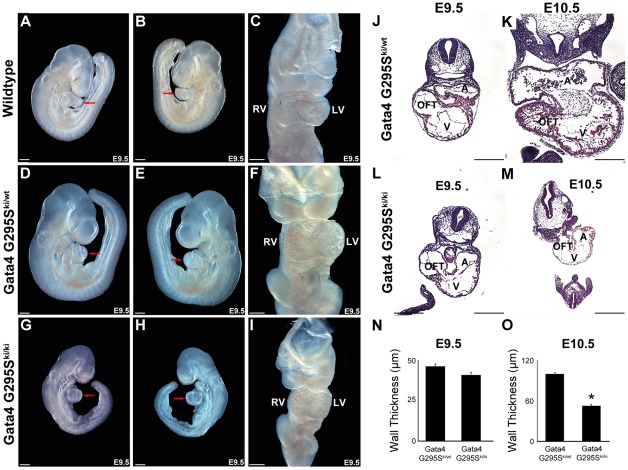
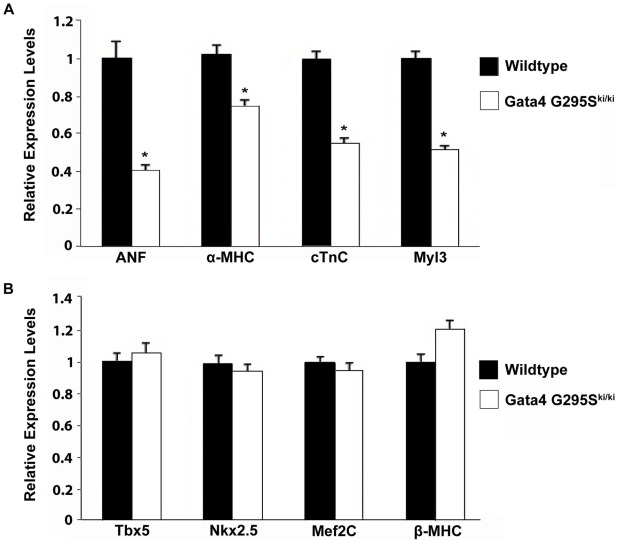
Center for Cardiovascular and Pulmonary Research and the Heart Center, Nationwide Children's Hospital, The Ohio State University, Columbus, Ohio, United States of America.
PLoS Genet. 2012;8(5):e1002690. doi: 10.1371/journal.pgen.1002690. Epub 2012 May 10.
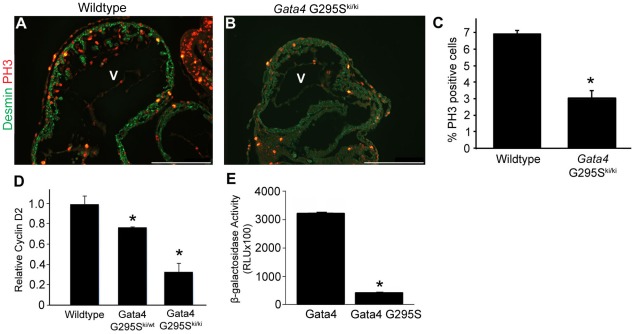
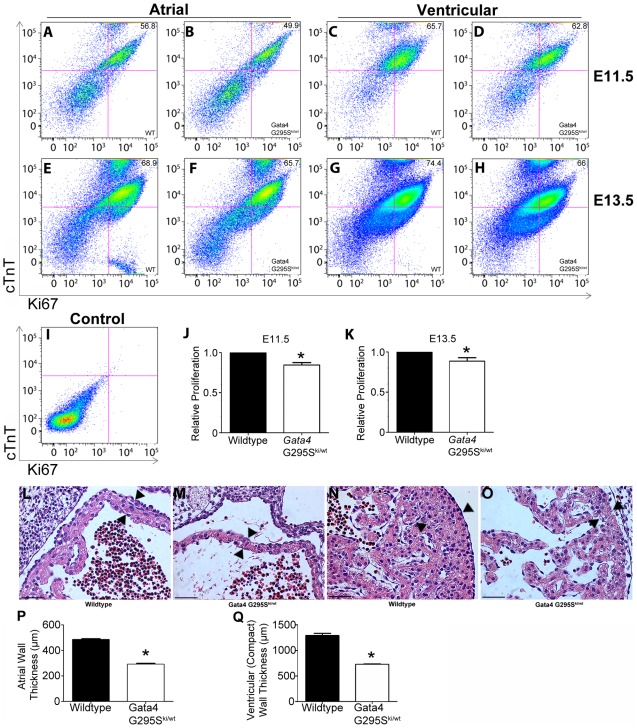
Defects of atrial and ventricular septation are the most frequent form of congenital heart disease, accounting for almost 50% of all cases. We previously reported that a heterozygous G296S missense mutation of GATA4 caused atrial and ventricular septal defects and pulmonary valve stenosis in humans. GATA4 encodes a cardiac transcription factor, and when deleted in mice it results in cardiac bifida and lethality by embryonic day (E)9.5. In vitro, the mutant GATA4 protein has a reduced DNA binding affinity and transcriptional activity and abolishes a physical interaction with TBX5, a transcription factor critical for normal heart formation. To characterize the mutation in vivo, we generated mice harboring the same mutation, Gata4 G295S. Mice homozygous for the Gata4 G295S mutant allele have normal ventral body patterning and heart looping, but have a thin ventricular myocardium, single ventricular chamber, and lethality by E11.5. While heterozygous Gata4 G295S mutant mice are viable, a subset of these mice have semilunar valve stenosis and small defects of the atrial septum. Gene expression studies of homozygous mutant mice suggest the G295S protein can sufficiently activate downstream targets of Gata4 in the endoderm but not in the developing heart. Cardiomyocyte proliferation deficits and decreased cardiac expression of CCND2, a member of the cyclin family and a direct target of Gata4, were found in embryos both homozygous and heterozygous for the Gata4 G295S allele. To further define functions of the Gata4 G295S mutation in vivo, compound mutant mice were generated in which specific cell lineages harbored both the Gata4 G295S mutant and Gata4 null alleles. Examination of these mice demonstrated that the Gata4 G295S protein has functional deficits in early myocardial development. In summary, the Gata4 G295S mutation functions as a hypomorph in vivo and leads to defects in cardiomyocyte proliferation during embryogenesis, which may contribute to the development of congenital heart defects in humans.
房间隔和室间隔缺损是最常见的先天性心脏病形式,占所有病例的近 50%。我们之前报道过,GATA4 的杂合 G296S 错义突变导致了人类的房间隔和室间隔缺损以及肺动脉瓣狭窄。GATA4 编码一种心脏转录因子,当在小鼠中缺失时,它会导致心脏裂和胚胎期第 9.5 天(E)的致死。在体外,突变的 GATA4 蛋白与 TBX5 的物理相互作用以及 DNA 结合亲和力和转录活性降低,而 TBX5 是正常心脏形成所必需的转录因子。为了在体内对突变进行特征描述,我们构建了携带相同突变(Gata4 G295S)的小鼠。纯合突变 Gata4 G295S 等位基因的小鼠具有正常的腹侧体模式和心脏环化,但心室心肌薄,单个心室腔,并在 E11.5 时致死。虽然杂合突变 Gata4 G295S 小鼠具有活力,但其中一部分具有半月瓣狭窄和房间隔小缺损。对纯合突变小鼠的基因表达研究表明,G295S 蛋白可以充分激活 Gata4 在内胚层中的下游靶标,但不能在发育中的心脏中激活。在 Gata4 G295S 等位基因纯合和杂合的胚胎中均发现了心肌细胞增殖缺陷和心脏 cyclin 家族成员 CCND2 的表达减少,CCND2 是 Gata4 的直接靶标。为了进一步确定 Gata4 G295S 突变在体内的功能,我们生成了携带 Gata4 G295S 突变和 Gata4 缺失等位基因的特定细胞谱系的复合突变小鼠。对这些小鼠的检查表明,Gata4 G295S 蛋白在早期心肌发育中具有功能缺陷。总之,Gata4 G295S 突变在体内作为一个次等位基因发挥作用,并导致胚胎发生期间心肌细胞增殖缺陷,这可能导致人类先天性心脏病的发展。