Research Division of Biology and Pathobiology of the Skin, Department of Dermatology, Medical University of Vienna, 1090 Vienna, Austria.
Allergy. 2013 Jan;68(1):37-47. doi: 10.1111/all.12051. Epub 2012 Nov 15.
Defects in keratinocyte differentiation and skin barrier are important features of inflammatory skin diseases like atopic dermatitis. Mast cells and their main mediator histamine are abundant in inflamed skin and thus may contribute to disease pathogenesis.
Human primary keratinocytes were cultured under differentiation-promoting conditions in the presence and absence of histamine, histamine receptor agonists and antagonists. The expression of differentiation-associated genes and epidermal junction proteins was quantified by real-time PCR, Western blot, and immunofluorescence labeling. The barrier function of human skin models was tested by the application of biotin as tracer molecule.
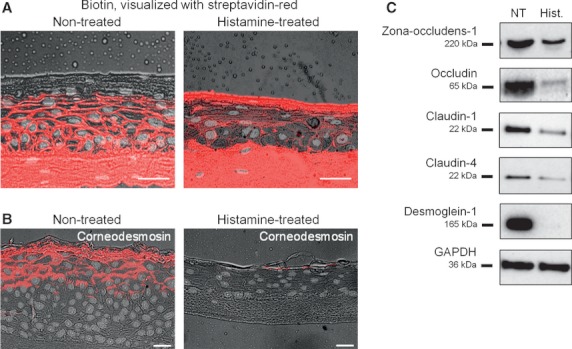
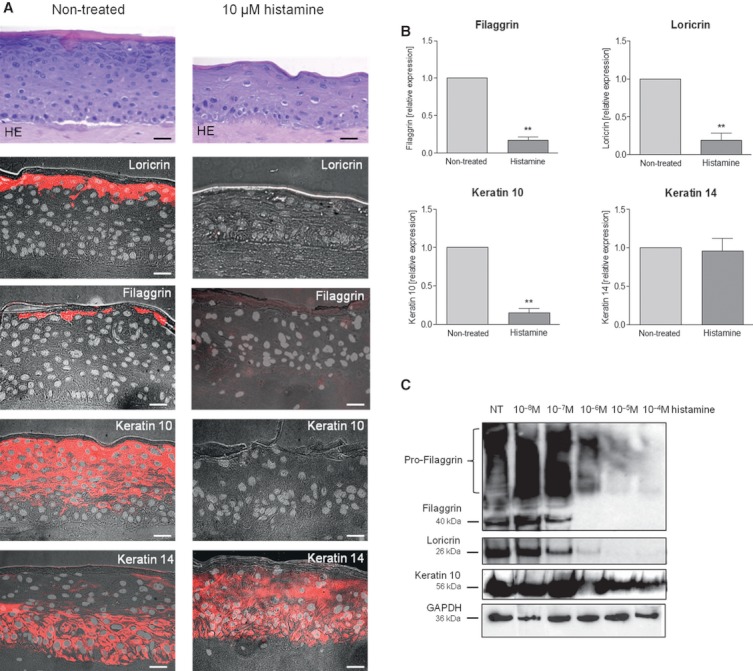
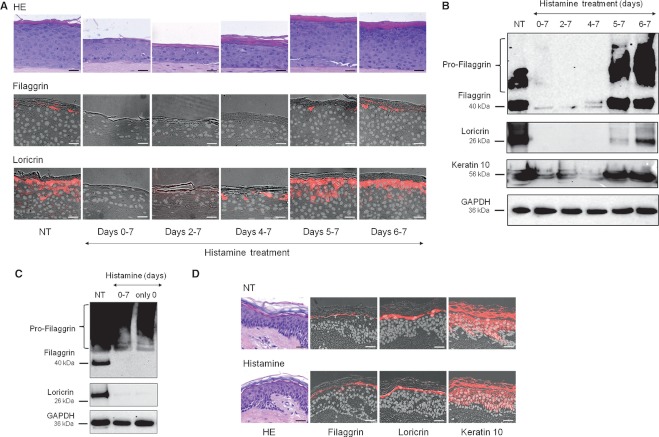
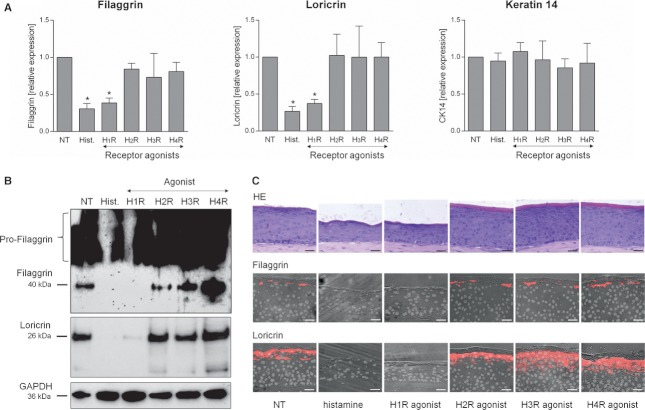
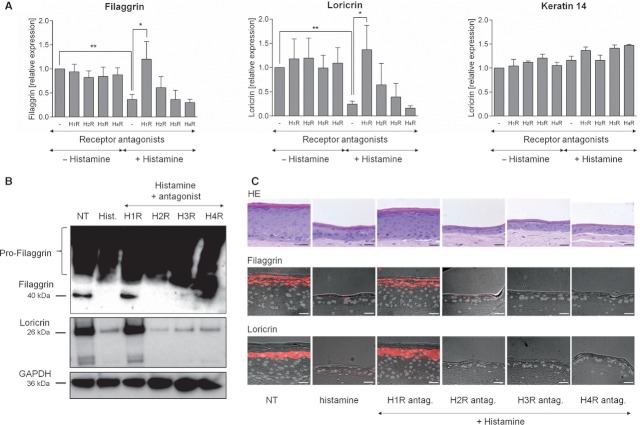
The addition of histamine to human keratinocyte cultures and organotypic skin models reduced the expression of the differentiation-associated proteins keratin 1/10, filaggrin, and loricrin by 80-95%. Moreover, the addition of histamine to skin models resulted in the loss of the granular layer and thinning of the epidermis and stratum corneum by 50%. The histamine receptor H1R agonist, 2-pyridylethylamine, suppressed keratinocyte differentiation to the same extent as did histamine. Correspondingly, cetirizine, an antagonist of H1R, virtually abrogated the effect of histamine. The expression of tight junction proteins zona occludens-1, occludin, claudin-1, and claudin-4, as well as that of desmosomal junction proteins corneodesmosin and desmoglein-1, was down-regulated by histamine. The tracer molecule biotin readily penetrated the tight junction barrier of skin cultures grown in the presence of histamine, while their diffusion was completely blocked in nontreated controls.
Our findings suggest a new mechanism by which mast cell activation and histamine release contribute to skin barrier defects in inflammatory skin diseases.
角蛋白细胞分化和皮肤屏障缺陷是特应性皮炎等炎症性皮肤病的重要特征。肥大细胞及其主要介质组胺在炎症皮肤中丰富,因此可能有助于疾病发病机制。
在存在和不存在组胺、组胺受体激动剂和拮抗剂的情况下,将人原代角质形成细胞在促进分化的条件下培养。通过实时 PCR、Western blot 和免疫荧光标记定量测定分化相关基因和表皮连接蛋白的表达。通过应用生物素作为示踪分子测试人皮肤模型的屏障功能。
将组胺添加到人角质形成细胞培养物和器官型皮肤模型中,使分化相关蛋白角蛋白 1/10、板层素和兜甲蛋白的表达减少 80-95%。此外,组胺添加到皮肤模型会导致颗粒层丢失和表皮和角质层变薄 50%。组胺受体 H1R 激动剂 2-吡啶乙胺与组胺一样抑制角质形成细胞分化。相应地,H1R 的拮抗剂西替利嗪实际上消除了组胺的作用。紧密连接蛋白 zonula occludens-1、occludin、claudin-1 和 claudin-4 的表达以及桥粒连接蛋白角蛋白丝和桥粒蛋白 1 的表达也受到组胺的下调。生物素示踪分子很容易穿透在组胺存在下生长的皮肤培养物的紧密连接屏障,而在未处理的对照中,其扩散完全被阻断。
我们的发现表明,肥大细胞活化和组胺释放通过新的机制导致炎症性皮肤病的皮肤屏障缺陷。