Department of Surgery, New York University Langone Medical Center, New York, NY, USA.
Hum Genet. 2013 May;132(5):523-36. doi: 10.1007/s00439-013-1269-4. Epub 2013 Jan 25.
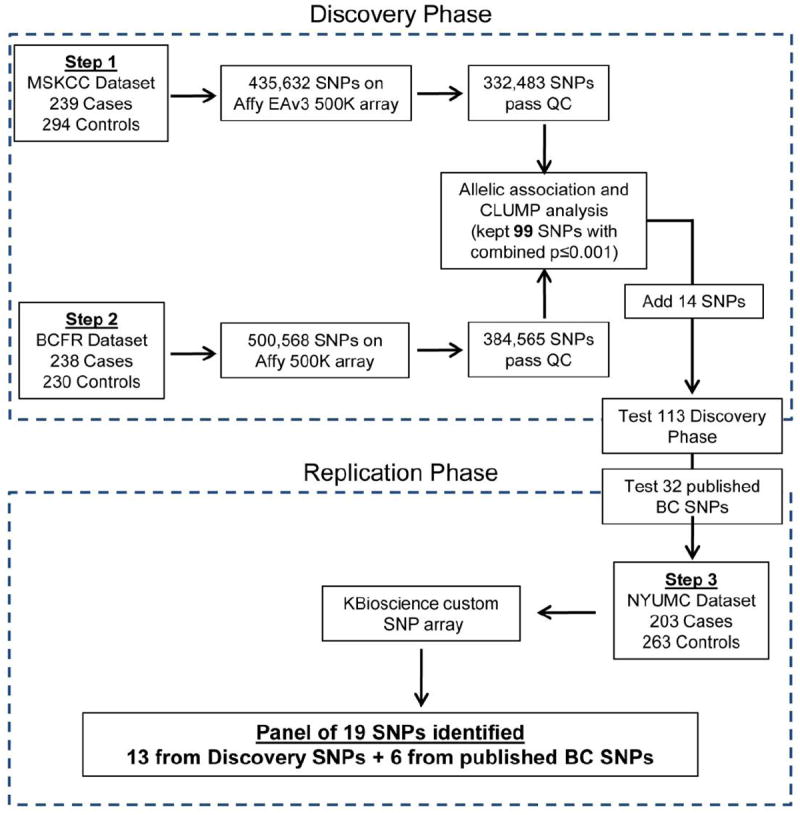
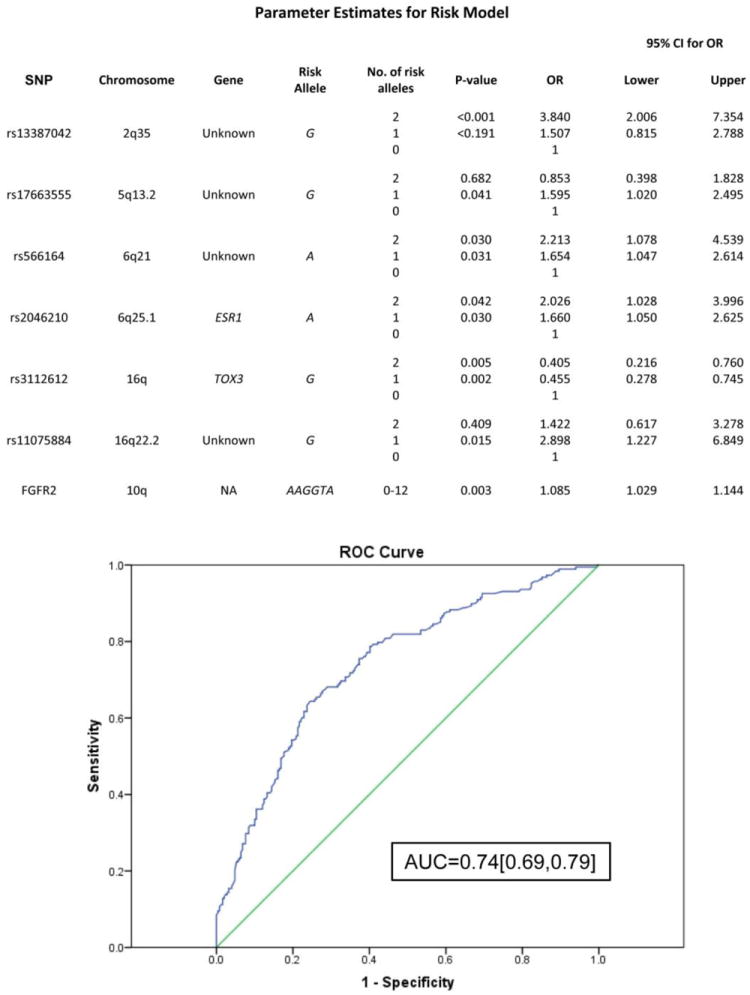
The ability to establish genetic risk models is critical for early identification and optimal treatment of breast cancer. For such a model to gain clinical utility, more variants must be identified beyond those discovered in previous genome-wide association studies (GWAS). This is especially true for women at high risk because of family history, but without BRCA1/2 mutations. This study incorporates three datasets in a GWAS analysis of women with Ashkenazi Jewish (AJ) homogeneous ancestry. Two independent discovery cohorts comprised 239 and 238 AJ women with invasive breast cancer or preinvasive ductal carcinoma in situ and strong family histories of breast cancer, but lacking the three BRCA1/2 founder mutations, along with 294 and 230 AJ controls, respectively. An independent, third cohort of 203 AJ cases with familial breast cancer history and 263 healthy controls of AJ women was used for validation. A total of 19 SNPs were identified as associated with familial breast cancer risk in AJ women. Among these SNPs, 13 were identified from a panel of 109 discovery SNPs, including an FGFR2 haplotype. In addition, six previously identified breast cancer GWAS SNPs were confirmed in this population. Seven of the 19 markers were significant in a multivariate predictive model of familial breast cancer in AJ women, three novel SNPs [rs17663555(5q13.2), rs566164(6q21), and rs11075884(16q22.2)], the FGFR2 haplotype, and three previously published SNPs [rs13387042(2q35), rs2046210(ESR1), and rs3112612(TOX3)], yielding moderate predictive power with an area under the curve (AUC) of the ROC (receiver-operator characteristic curve) of 0.74. Population-specific genetic variants in addition to variants shared with populations of European ancestry may improve breast cancer risk prediction among AJ women from high-risk families without founder BRCA1/2 mutations.
建立遗传风险模型的能力对于乳腺癌的早期识别和最佳治疗至关重要。为了使这种模型具有临床实用性,必须在以前的全基因组关联研究(GWAS)中发现的变异之外,发现更多的变异。对于那些由于家族史而处于高风险但没有 BRCA1/2 突变的女性来说尤其如此。本研究在对具有阿什肯纳兹犹太(AJ)同质血统的女性进行 GWAS 分析时,结合了三个数据集。两个独立的发现队列由 239 名和 238 名 AJ 女性组成,她们患有浸润性乳腺癌或导管内原位癌,并且有强烈的乳腺癌家族史,但缺乏三种 BRCA1/2 创始人突变,分别与 294 名和 230 名 AJ 对照者相匹配。一个独立的、第三个由 203 名 AJ 病例组成的队列,这些病例具有家族性乳腺癌病史,263 名 AJ 女性健康对照者用于验证。总共确定了 19 个 SNP 与 AJ 女性的家族性乳腺癌风险相关。在这些 SNP 中,有 13 个是从 109 个发现 SNP 的面板中鉴定出来的,其中包括一个 FGFR2 单倍型。此外,在该人群中还确认了六个先前确定的乳腺癌 GWAS SNP。在 AJ 女性的家族性乳腺癌多变量预测模型中,有 7 个标记物具有统计学意义,其中 3 个是新的 SNP [rs17663555(5q13.2)、rs566164(6q21)和 rs11075884(16q22.2)]、FGFR2 单倍型和三个先前发表的 SNP [rs13387042(2q35)、rs2046210(ESR1)和 rs3112612(TOX3)],ROC(接收器操作特征曲线)的 AUC 为 0.74,具有中等预测能力。除了与欧洲血统人群共享的变异外,特定于人群的遗传变异可能会提高没有创始人 BRCA1/2 突变的 AJ 高危家族女性的乳腺癌风险预测。