Friedrich-Baur-Institute, Department of Neurology, Ludwig-Maximilians-University of Munich, Ziemssenstr. 1a, Munich 80336, Germany.
Orphanet J Rare Dis. 2013 Feb 14;8:26. doi: 10.1186/1750-1172-8-26.
Dysferlinopathies are autosomal recessive disorders caused by mutations in the dysferlin (DYSF) gene encoding the dysferlin protein. DYSF mutations lead to a wide range of muscular phenotypes, with the most prominent being Miyoshi myopathy (MM) and limb girdle muscular dystrophy type 2B (LGMD2B).
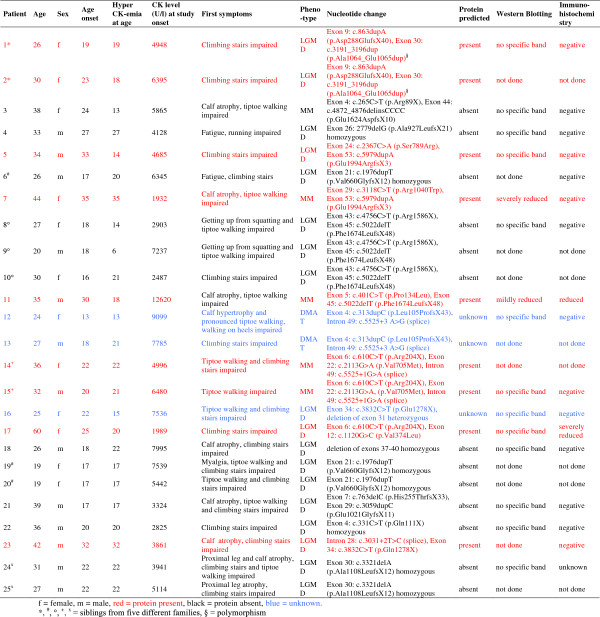
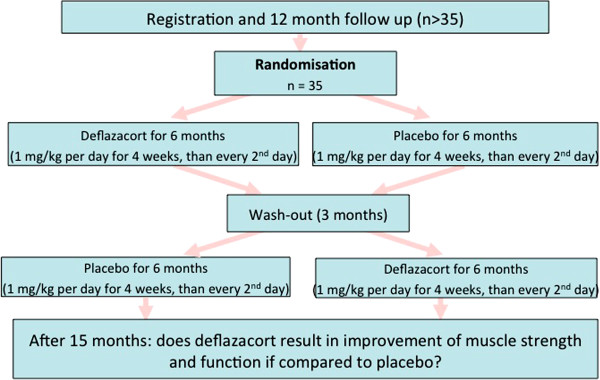
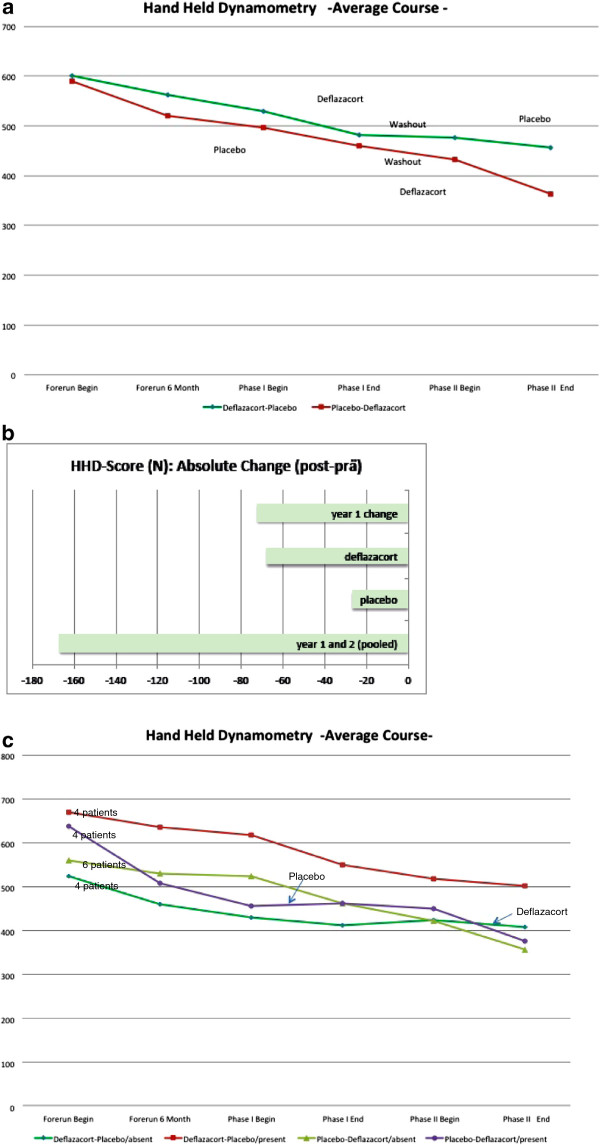
We assessed the one-year-natural course of dysferlinopathy, and the safety and efficacy of deflazacort treatment in a double-blind, placebo-controlled cross-over trial. After one year of natural course without intervention, 25 patients with genetically defined dysferlinopathy were randomized to receive deflazacort and placebo for six months each (1 mg/kg/day in month one, 1 mg/kg every 2nd day during months two to six) in one of two treatment sequences.
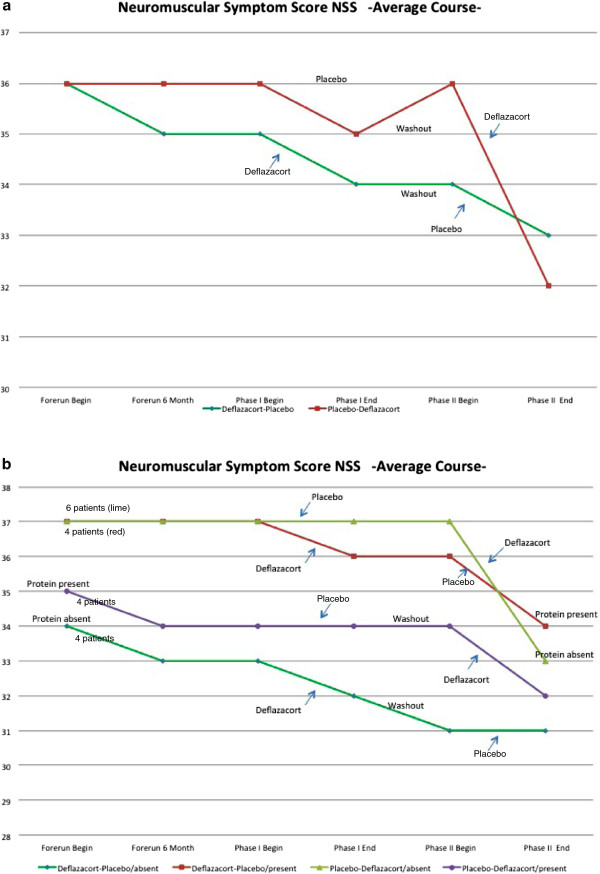
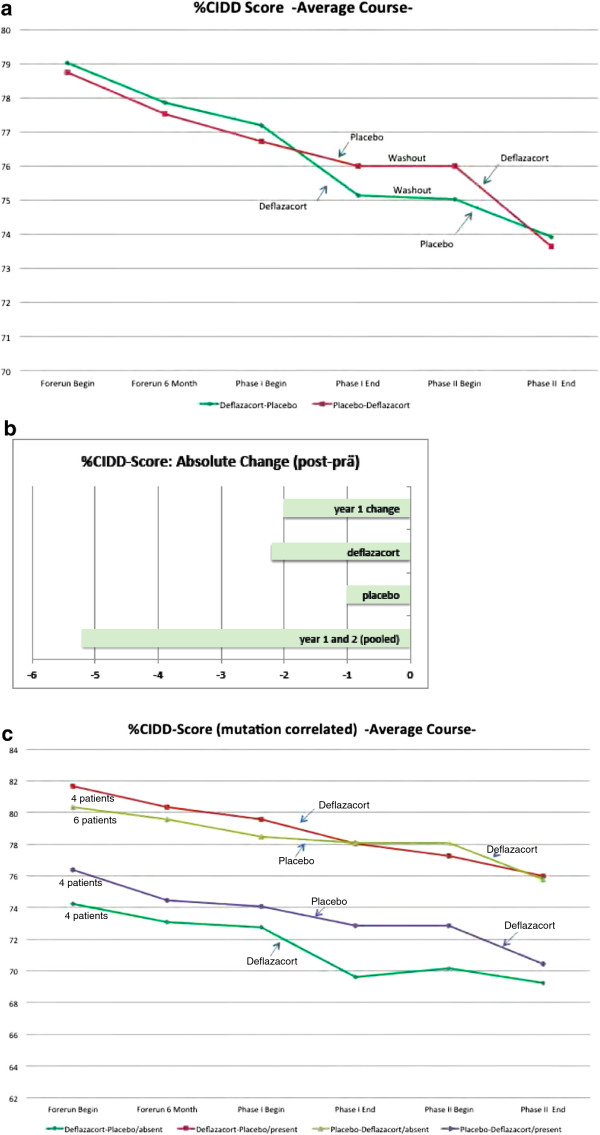
During one year of natural course, muscle strength declined about 2% as measured by CIDD (Clinical Investigation of Duchenne Dystrophy) score, and 76 Newton as measured by hand-held dynamometry. Deflazacort did not improve muscle strength. In contrast, there is a trend of worsening muscle strength under deflazacort treatment, which recovers after discontinuation of the study drug. During deflazacort treatment, patients showed a broad spectrum of steroid side effects.
Deflazacort is not an effective therapy for dysferlinopathies, and off-label use is not warranted. This is an important finding, since steroid treatment should not be administered in patients with dysferlinopathy, who may be often misdiagnosed as polymyositis.
This clinical trial was registered at http://www.ClincalTrials.gov, identifier: NCT00527228, and was always freely accessible to the public.
肌营养不良蛋白病是由肌营养不良蛋白(DYSF)基因突变引起的常染色体隐性遗传病。DYSF 基因突变导致广泛的肌肉表型,其中最突出的是 Miyoshi 肌病(MM)和肢带型肌营养不良 2B 型(LGMD2B)。
我们评估了肌营养不良蛋白病的一年自然病程,以及在一项双盲、安慰剂对照交叉试验中使用地夫可特治疗的安全性和疗效。在未经干预的一年自然病程后,25 名遗传性肌营养不良蛋白病患者被随机分为两组,分别接受地夫可特和安慰剂治疗 6 个月(第 1 个月 1mg/kg/天,第 2 至 6 个月 1mg/kg/每 2 天),两种治疗方案之一。
在一年的自然病程中,肌肉力量下降了约 2%,通过 CIDD(Duchenne 肌营养不良症临床研究)评分和 76 牛顿通过手持测力计测量。地夫可特并不能改善肌肉力量。相反,在接受地夫可特治疗时,肌肉力量有恶化的趋势,在停药后恢复。在接受地夫可特治疗期间,患者出现了一系列类固醇副作用。
地夫可特不是肌营养不良蛋白病的有效治疗方法,不应该进行标签外使用。这是一个重要的发现,因为类固醇治疗不应在肌营养不良蛋白病患者中进行,这些患者常常被误诊为多发性肌炎。
该临床试验在 http://www.clincaltrials.gov 上注册,标识符:NCT00527228,始终向公众免费开放。