1Bristol Heart Institute & School of Cellular and Molecular Medicine,University of Bristol, Bristol, UK.
BMC Med Genet. 2013 Apr 4;14:42. doi: 10.1186/1471-2350-14-42.
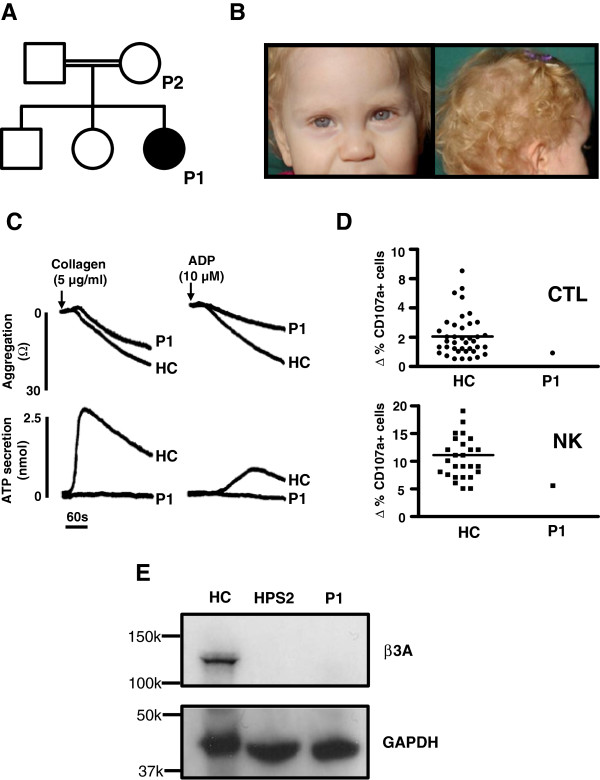
Hermansky-Pudlak syndrome 2 (HPS2; OMIM #608233) is a rare, autosomal recessive disorder caused by loss-of-function genetic variations affecting AP3B1, which encodes the β3A subunit of the adaptor-related protein complex 3 (AP3). Phenotypic characteristics include reduced pigmentation, absent platelet dense granule secretion, neutropenia and reduced cytotoxic T lymphocyte (CTL) and natural killer (NK) cell function. To date HPS2 has been associated with non-synonymous, stop-gain or deletion-insertion nucleotide variations within the coding region of AP3B1.
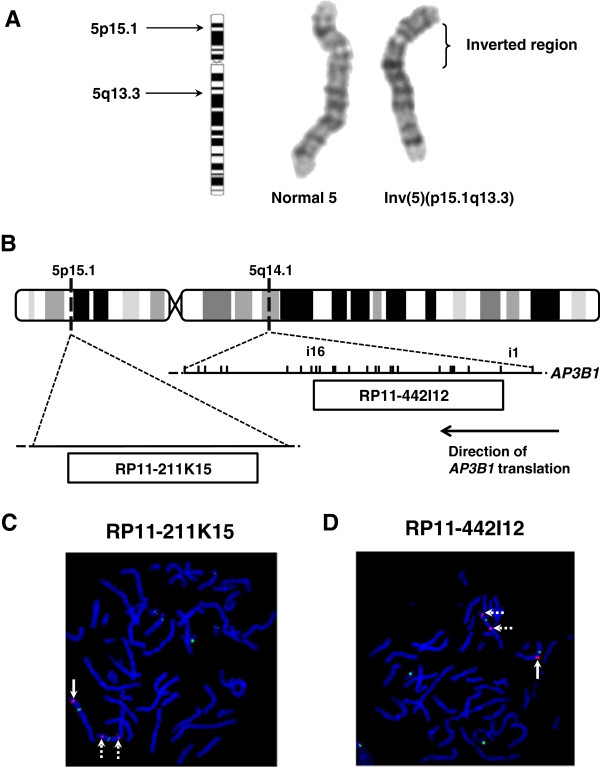
We describe a consanguineous female infant with reduced pigmentation, neutropenia and recurrent infections. Platelets displayed reduced aggregation and absent ATP secretion in response to collagen and ADP, indicating a platelet dense granule defect. There was increased basal surface expression of CD107a (lysosome-associated membrane protein 1(LAMP-1)) on NK cells and CTLs from the study subject and a smaller increase in the percentage of CD107a positive cells after stimulation compared to most healthy controls. Immunoblotting of protein extracts from EBV-transformed lymphoblasts from the index case showed absent expression of full-length AP-3 β3A subunit protein, confirming a phenotypic diagnosis of HPS2.The index case displayed a homozygous pericentric inv(5)(p15.1q14.1), which was also detected as a heterozygous defect in both parents of the index case. No loss of genetic material was demonstrated by microarray comparative genome hybridisation at 60kb resolution. Fluorescence in-situ hybridisation using the 189.6kb probe RP11-422I12, which maps to 5q14.1, demonstrated dual hybridisation to both 5q14.1 and 5p15.1 regions of the inverted Chr5. The RP11-422I12 probe maps from intron 1 to intron 16 of AP3B1, thus localising the 5q inversion breakpoint to within AP3B1. The probe RP11-211K15, which corresponds to an intergenic region on 5p also showed dual hybridisation, enabling localisation of the 5p inversion breakpoint.
This case report extends the phenotypic description of the very rare disorder HPS2. Our demonstration of a homozygous Chr5 inversion predicted to disrupt AP3B1 gene provides a novel pathogenic mechanism for this disorder.
Hermansky-Pudlak 综合征 2 型(HPS2;OMIM#608233)是一种罕见的常染色体隐性遗传疾病,由影响 AP3B1 的功能丧失基因突变引起,AP3B1 编码衔接相关蛋白复合物 3(AP3)的β3A 亚基。表型特征包括色素减退、血小板致密颗粒分泌缺失、中性粒细胞减少以及细胞毒性 T 淋巴细胞(CTL)和自然杀伤(NK)细胞功能降低。迄今为止,HPS2 与 AP3B1 编码区的非同义、无义突变或缺失-插入核苷酸变异有关。
我们描述了一名有色素减退、中性粒细胞减少和反复感染的同系女婴。血小板对胶原和 ADP 的聚集反应减弱,ATP 分泌缺失,提示血小板致密颗粒缺陷。与大多数健康对照者相比,研究对象的 NK 细胞和 CTL 细胞的基础表面 CD107a(溶酶体相关膜蛋白 1(LAMP-1))表达增加,刺激后 CD107a 阳性细胞的百分比也略有增加。对该指数病例的 EBV 转化的淋巴母细胞蛋白提取物进行免疫印迹分析显示,全长 AP-3β3A 亚基蛋白表达缺失,证实了 HPS2 的表型诊断。该指数病例表现出纯合性中央着丝粒 inv(5)(p15.1q14.1),在指数病例的父母中也检测到杂合性缺陷。在分辨率为 60kb 的微阵列比较基因组杂交中未显示遗传物质缺失。使用映射到 5q14.1 的 189.6kb 探针 RP11-422I12 的荧光原位杂交显示,两个 5q14.1 和 5p15.1 区域的 Chr5 倒置均发生杂交。RP11-422I12 探针从 AP3B1 的内含子 1 到内含子 16 映射,因此将 5q 倒置断点定位在 AP3B1 内。对应于 5p 上的一个基因间区域的探针 RP11-211K15 也显示出双重杂交,从而使 5p 倒置断点定位。
本病例报告扩展了非常罕见的 HPS2 疾病的表型描述。我们证明了一种纯合性 Chr5 倒置,预计会破坏 AP3B1 基因,为这种疾病提供了一种新的致病机制。