Institute for Science and Technology in Medicine, Keele University, Huxley Building, Staffordshire ST5 5BG, United Kingdom.
BMC Genomics. 2013 Apr 19;14:267. doi: 10.1186/1471-2164-14-267.
The ability of the human malarial parasite Plasmodium falciparum to invade, colonise and multiply within diverse host environments, as well as to manifest its virulence within the human host, are activities tightly linked to the temporal and spatial control of gene expression. Yet, despite the wealth of high throughput transcriptomic data available for this organism there is very little information regarding the location of key transcriptional landmarks or their associated cis-acting regulatory elements. Here we provide a systematic exploration of the size and organisation of transcripts within intergenic regions to yield surrogate information regarding transcriptional landmarks, and to also explore the spatial and temporal organisation of transcripts over these poorly characterised genomic regions.
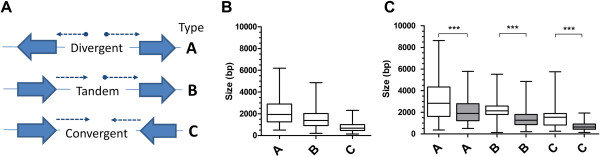
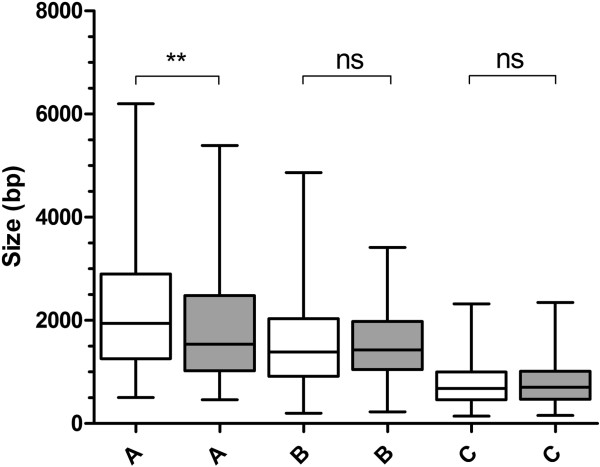
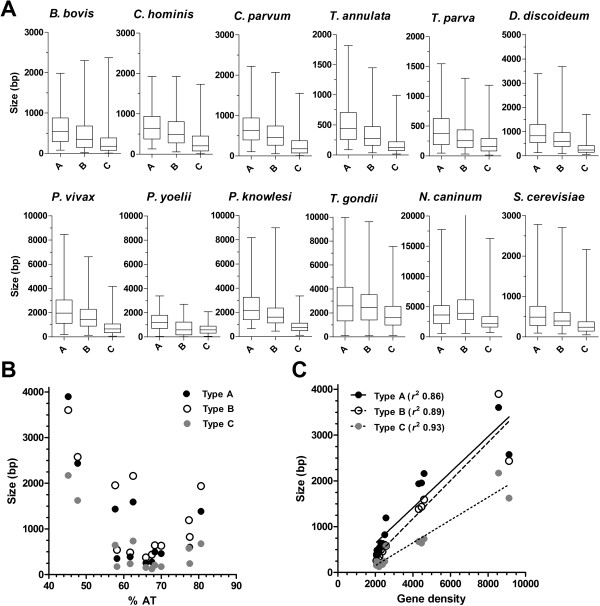
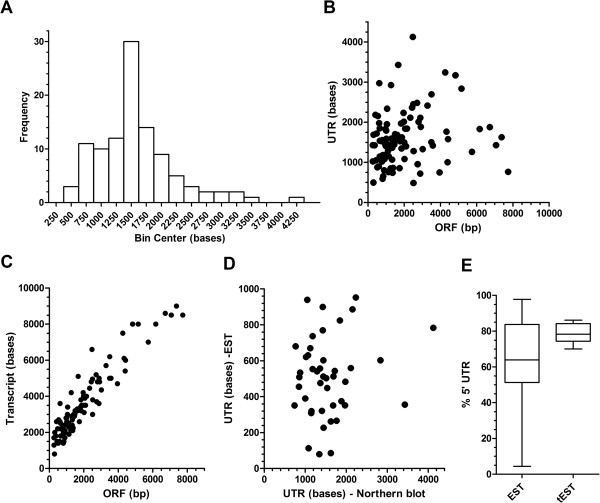
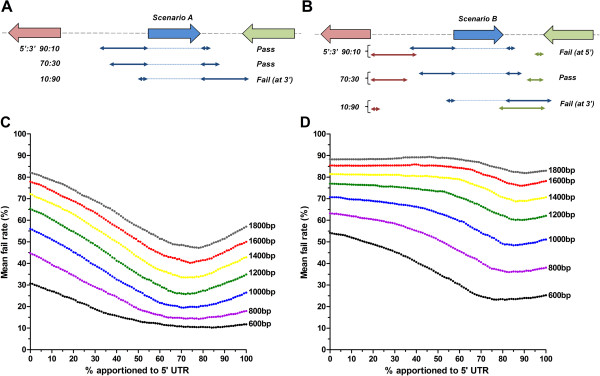
Utilising the transcript data for a cohort of 105 genes we demonstrate that the untranscribed regions of mRNA are large and apportioned predominantly to the 5' end of the open reading frame. Given the relatively compact size of the P. falciparum genome, we suggest that whilst transcriptional units are likely to spatially overlap, temporal co-transcription of adjacent transcriptional units is actually limited. Critically, the size of intergenic regions is directly dependent on the orientation of the two transcriptional units arrayed over them, an observation we extend to an analysis of the complete sequences of twelve additional organisms that share moderately compact genomes.
Our study provides a theoretical framework that extends our current understanding of the transcriptional landscape across the P. falciparum genome. Demonstration of a consensus gene-spacing rule that is shared between P. falciparum and ten other moderately compact genomes of apicomplexan parasites reveals the potential for our findings to have a wider impact across a phylum that contains many organisms important to human and veterinary health.
人类疟原虫恶性疟原虫能够在不同的宿主环境中入侵、定殖和繁殖,以及在人类宿主中表现出其毒力,这些活动与基因表达的时空控制密切相关。然而,尽管有大量高通量转录组数据可用于该生物,但关于关键转录起始位点的位置或其相关顺式作用调节元件的信息却非常有限。在这里,我们系统地研究了基因间区转录本的大小和组织,以提供关于转录起始位点的替代信息,并探索这些 poorly 表征的基因组区域中转录本的时空组织。
利用 105 个基因的转录组数据,我们证明了 mRNA 的非转录区很大,主要分配到开放阅读框的 5'端。鉴于恶性疟原虫基因组相对紧凑的大小,我们认为虽然转录单元可能在空间上重叠,但相邻转录单元的共转录实际上是有限的。至关重要的是,基因间区的大小直接取决于排列在其上的两个转录单元的方向,我们将这一观察结果扩展到对另外 12 种具有中度紧凑基因组的生物体的完整序列的分析。
我们的研究提供了一个理论框架,扩展了我们对恶性疟原虫基因组中转录景观的现有理解。在恶性疟原虫和其他十种中度紧凑的顶复门寄生虫基因组之间共享的一致基因间隔规则的证明揭示了我们的发现可能在包含许多对人类和兽医健康重要的生物体的门中产生更广泛的影响的潜力。