Department of Biological Sciences, University of Maryland, Baltimore County, Baltimore, MD, USA.
BMC Genomics. 2013;14 Suppl 3(Suppl 3):S5. doi: 10.1186/1471-2164-14-S3-S5. Epub 2013 May 28.
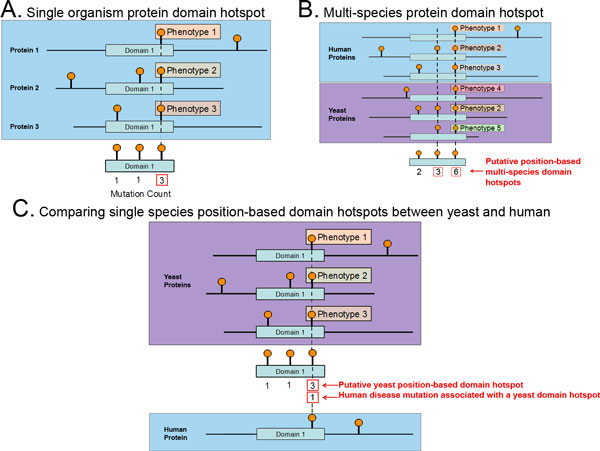
The body of disease mutations with known phenotypic relevance continues to increase and is expected to do so even faster with the advent of new experimental techniques such as whole-genome sequencing coupled with disease association studies. However, genomic association studies are limited by the molecular complexity of the phenotype being studied and the population size needed to have adequate statistical power. One way to circumvent this problem, which is critical for the study of rare diseases, is to study the molecular patterns emerging from functional studies of existing disease mutations. Current gene-centric analyses to study mutations in coding regions are limited by their inability to account for the functional modularity of the protein. Previous studies of the functional patterns of known human disease mutations have shown a significant tendency to cluster at protein domain positions, namely position-based domain hotspots of disease mutations. However, the limited number of known disease mutations remains the main factor hindering the advancement of mutation studies at a functional level. In this paper, we address this problem by incorporating mutations known to be disruptive of phenotypes in other species. Focusing on two evolutionarily distant organisms, human and yeast, we describe the first inter-species analysis of mutations of phenotypic relevance at the protein domain level.
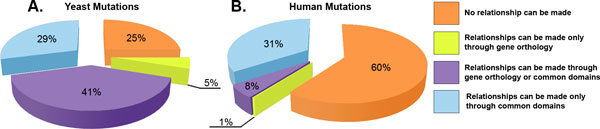
The results of this analysis reveal that phenotypic mutations from yeast cluster at specific positions on protein domains, a characteristic previously revealed to be displayed by human disease mutations. We found over one hundred domain hotspots in yeast with approximately 50% in the exact same domain position as known human disease mutations.
We describe an analysis using protein domains as a framework for transferring functional information by studying domain hotspots in human and yeast and relating phenotypic changes in yeast to diseases in human. This first-of-a-kind study of phenotypically relevant yeast mutations in relation to human disease mutations demonstrates the utility of a multi-species analysis for advancing the understanding of the relationship between genetic mutations and phenotypic changes at the organismal level.
具有已知表型相关性的疾病突变体数量不断增加,随着全基因组测序与疾病关联研究等新实验技术的出现,预计这一趋势还会加快。然而,基因组关联研究受到所研究表型的分子复杂性以及获得足够统计能力所需的群体规模的限制。对于罕见病的研究来说,规避这一问题的一种方法是研究现有疾病突变的功能研究中出现的分子模式。目前,针对编码区域突变的基于基因的分析受到其无法解释蛋白质功能模块化的限制。先前对已知人类疾病突变的功能模式的研究表明,突变显著倾向于聚集在蛋白质结构域位置,即疾病突变的基于位置的结构域热点。然而,已知疾病突变的数量有限仍然是阻碍功能水平突变研究进展的主要因素。在本文中,我们通过纳入已知在其他物种中导致表型破坏的突变来解决这一问题。我们专注于两个进化上相距甚远的生物体,人类和酵母,描述了蛋白质结构域水平上第一个具有表型相关性的物种间突变的分析。
该分析的结果表明,来自酵母的表型突变聚集在蛋白质结构域的特定位置,这一特征先前已被揭示出是人类疾病突变所具有的。我们在酵母中发现了一百多个结构域热点,其中约有 50%位于与已知人类疾病突变相同的结构域位置。
我们描述了一种使用蛋白质结构域作为框架的分析方法,通过研究人类和酵母中的结构域热点并将酵母中的表型变化与人类疾病联系起来,来转移功能信息。这项首次针对与人类疾病相关的具有表型相关性的酵母突变的研究表明,多物种分析对于推进对遗传突变与生物体水平表型变化之间关系的理解具有实用性。