Department of Chemical and Systems Biology, Stanford University, Stanford, California, United States of America.
PLoS Biol. 2013 Jul;11(7):e1001599. doi: 10.1371/journal.pbio.1001599. Epub 2013 Jul 2.
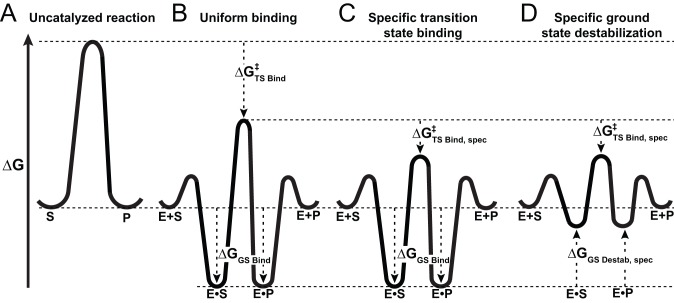
Enzymes stabilize transition states of reactions while limiting binding to ground states, as is generally required for any catalyst. Alkaline Phosphatase (AP) and other nonspecific phosphatases are some of Nature's most impressive catalysts, achieving preferential transition state over ground state stabilization of more than 10²²-fold while utilizing interactions with only the five atoms attached to the transferred phosphorus. We tested a model that AP achieves a portion of this preference by destabilizing ground state binding via charge repulsion between the anionic active site nucleophile, Ser102, and the negatively charged phosphate monoester substrate. Removal of the Ser102 alkoxide by mutation to glycine or alanine increases the observed Pi affinity by orders of magnitude at pH 8.0. To allow precise and quantitative comparisons, the ionic form of bound P(i) was determined from pH dependencies of the binding of Pi and tungstate, a P(i) analog lacking titratable protons over the pH range of 5-11, and from the ³¹P chemical shift of bound P(i). The results show that the Pi trianion binds with an exceptionally strong femtomolar affinity in the absence of Ser102, show that its binding is destabilized by ≥10⁸-fold by the Ser102 alkoxide, and provide direct evidence for ground state destabilization. Comparisons of X-ray crystal structures of AP with and without Ser102 reveal the same active site and P(i) binding geometry upon removal of Ser102, suggesting that the destabilization does not result from a major structural rearrangement upon mutation of Ser102. Analogous Pi binding measurements with a protein tyrosine phosphatase suggest the generality of this ground state destabilization mechanism. Our results have uncovered an important contribution of anionic nucleophiles to phosphoryl transfer catalysis via ground state electrostatic destabilization and an enormous capacity of the AP active site for specific and strong recognition of the phosphoryl group in the transition state.
酶稳定反应的过渡态,同时限制与基态的结合,这是任何催化剂通常所需要的。碱性磷酸酶(AP)和其他非特异性磷酸酶是自然界中最令人印象深刻的催化剂之一,它们通过与转移磷原子相连的仅五个原子的相互作用,实现了超过 10²²倍的过渡态优先稳定,而不是基态稳定。我们测试了一个模型,即 AP 通过带负电荷的活性位点亲核试剂 Ser102 与带负电荷的磷酸单酯底物之间的电荷排斥作用,使基态结合不稳定,从而实现了这种偏好的一部分。通过突变将 Ser102 的烷氧基转化为甘氨酸或丙氨酸,可使 pH 8.0 时观察到的 Pi 亲和力增加几个数量级。为了进行精确和定量的比较,从 Pi 和钨酸盐(一种在 pH 5-11 范围内没有可滴定质子的 Pi 类似物)结合的 pH 依赖性以及结合 Pi 的 ³¹P 化学位移确定了结合的 P(i) 的离子形式。结果表明,在没有 Ser102 的情况下,Pi 三阴离子以异常强的飞摩尔亲和力结合,表明其结合被 Ser102 烷氧基稳定化 ≥10⁸倍,并提供了对基态失稳的直接证据。与含有和不含 Ser102 的 AP 的 X 射线晶体结构的比较表明,在 Ser102 缺失后,其活性位点和 P(i)结合几何形状相同,这表明这种失稳不是由于 Ser102 突变后发生了重大的结构重排。与蛋白酪氨酸磷酸酶的类似的 Pi 结合测量表明,这种基态失稳机制具有普遍性。我们的研究结果揭示了带负电荷的亲核试剂通过基态静电失稳对磷酸转移催化的重要贡献,以及 AP 活性位点对过渡态中磷酸基团的特异性和强识别的巨大能力。